
The Collaborative Research Center 1530 (SFB1530) is a network of internationally visible investigators with highly complementary technical expertise in innovative lymphoma models, cutting-edge genomics and proteomics platforms, comprehensively characterized, large cohorts of lymphoma patients, powerful bioinformatics and a successful clinical trial infrastructure.
The SFB1530 is specifically designed to create a cooperation platform of clinicians and scientists with excellent track-records either in lymphoma or in inflammation research. In doing so, the overarching goal of the Collaborative Research Center is to create and strengthen synergies towards the discovery of patho-mechanisms and the identification of innovative therapeutic strategies beyond the lymphoma cell-autonomous level. The close interactions with oncology centers of excellence in Cologne, Essen, Frankfurt, Berlin and Heidelberg and with major German lymphoma and leukemia study groups will allow for a rapid translation of major findings into clinical application.
In summary, the SFB1530 will undertake a holistic and interdisciplinary effort, which will create a comprehensive inventory of the major patho-mechanisms that define high-risk B cell malignancies in their specific microenvironment as a molecular framework for mechanism-based therapies.
News

Women & Science: Advancing Cancer Research - Elisa ten Hacken
Join our first seminar in the Women & Science series

Seminar: Early Career Researchers in Oncology - funding opportunities
Date: Wednesday, May 7 2024, 10:00 am - 12:00 pm
Location: University Hospital…

SFB 1530 Seminar Series: Michael Hudecek
Our next seminar will be held by Michael Hudecek, chair of the Department for…

Workshop: Research Data Management in (Bio-) Medicine
Register for the first workshop on RDM in an upcoming series

Else Kröner Excellence Stipend for Paul Bröckelmann
Congrats to Paul on this achievement!
New publication by Prinz, Chmielewski et al.: new strategy for safer CAR T Cell therapy in lymphomas
The preclinical study reveals promising strategies for translational research
New publication: Role of the tumor microenvironment in CLL pathogenesis
New review by project B01 in press (pre-proof)
New publication by Al-Sawaf, Eichhorst et al. in Nature Medicine
The phase 2 RT1 trial shows that deep and durable remissions can be achieved in…

SFB 1530 Seminar Series: Philipp Jost
Our next seminar will be held by Philipp Jost, professor in the Division of…
Call for Abstracts: West German Lymphoma Symposium
The bi-annual West German Lymphoma Symposium launches in May 2024

SFB 1530 Seminar Series: Tríona Ní Chonghaile
Dr Tríona Ní Chonghaile is a senior lecturer in the Department of Physiology and…

Bridging Perspectives: A Dialogue on Diversity and Mental Health in Science
EXHIBITION from November 16th to December 1st, 2023

It's a wrap – annual retreat in Estoril
Our second retreat took place 22 and 23 October 2023 in Estoril, Portugal
New publication by Kashkar et al. in The EMBO Journal
Abstract
Cellular inhibitor of apoptosis proteins (cIAPs) are RING-containing…

SFB 1530 Seminar Series: Arnon Kater
Arnon Kater's research covers the broad spectrum of fundamental, pre-clinical…
New publication in Science Advances: cFLIP and cell death
The Annibaldi lab's study on the role of cFLIP cleavage in the regulation of…
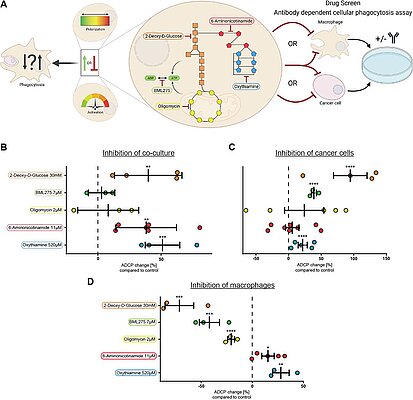
New preprint of the Pallasch lab: Pentose Phosphate Pathway Inhibition activates Macrophages towards phagocytic Lymphoma Cell Clearance
Macrophages in the B-cell lymphoma microenvironment represent a functional node…

SFB 1530 Seminar Series: Caroline Pabst (Heidelberg University Hospital)
About Caroline Pabst, MD
Caroline Pabst is a clinician researcher at Heidelberg…

SFB 1530 Diversity Coffee Break
Take a coffee break with the Inclusivity, Diversity and Gender committee!
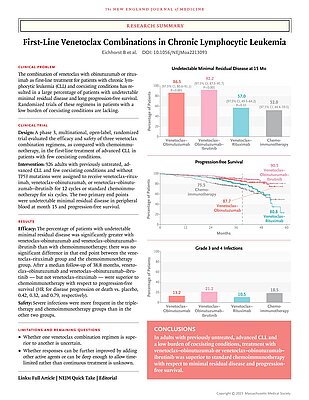
New publication in The New England Journal of Medicine
The latest study of the German CLL Study Group shows that…

SFB 1530 Seminar Series kickoff: Martin Dreyling (LMU Munich)
Our first guest speaker for the series is Prof. Martin Dreyling from LMU Munich.
…
New publication in Nature Communications
New publication of project C04 in B-cell lymphoma and CLL: Transcriptomic…

Christian Reinhardt receives the German Cancer Award 2023
The CRC1530 group leader was awarded the German Cancer Award 2023 in…
Call for Abstracts/Registration: 7th Translational Research Conference: Lymphoid Malignancies
The European School of Haematology (ESH) 7th Translational Research Conference…
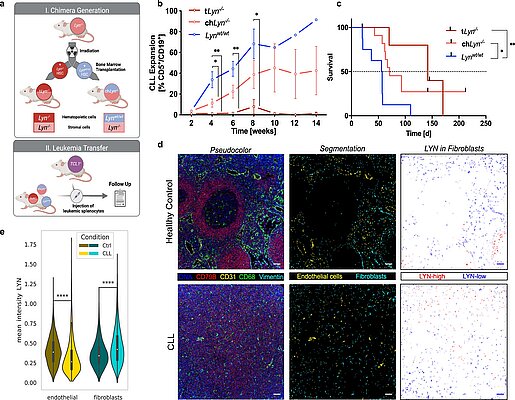
New Publication: LYN kinase programs stromal fibroblasts
The new publication by Phuong-Hien Nguyen and Michael Hallek's team in Nature…
Latest Publications
-
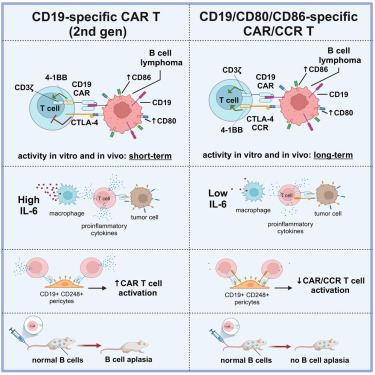
An anti-CD19/CTLA-4 switch improves efficacy and selectivity of CAR T cells targeting CD80/86-upregulated DLBCL
himeric antigen receptor T cell (CAR T) therapy is a potent treatment for relapsed/refractory (r/r) B cell lymphomas but provides lasting remissions in only ∼40% of patients and is associated with serious adverse events. We identify an upregulation of CD80 and/or CD86 in tumor tissue of (r/r) diffuse large B cell lymphoma (DLBCL) patients treated with tisagenlecleucel. This finding leads to the development of the CAR/CCR (chimeric checkpoint receptor) design, which consists of a CD19-specific first-generation CAR co-expressed with a recombinant CTLA-4-linked receptor with a 4-1BB co-stimulatory domain. CAR/CCR T cells demonstrate superior efficacy in xenograft mouse models compared with CAR T cells, superior long-term activity, and superior selectivity in in vitro assays with non-malignant CD19+ cells. In addition, immunocompetent mice show an intact CD80−CD19+ B cell population after CAR/CCR T cell treatment. The results reveal the CAR/CCR design as a promising strategy for further translational study.
-
Noninvasive minimal residual disease assessment in relapsed/refractory large B-cell lymphoma using digital droplet PCR
Although several promising approaches for the treatment of relapsed/refractory diffuse large B-cell lymphoma (rrDLBCL) have been approved recently, it remains unclear which patients will ultimately achieve long-term responses. Circulating tumor (ct)DNA sequencing has emerged as a valuable tool to assess minimal residual disease (MRD). Correlations between MRD and outcomes have been shown in previously untreated DLBCL, but data on the repeated assessment of MRD in the dynamic course of rrDLBCL is limited. Here, we present an approach leveraging cost- and time-sensitivity of digital droplet (dd)PCR to repeatedly assess MRD in rrDLBCL and present proof-of-principle for its ability to predict outcomes.
-
Entirely noninvasive outcome prediction in central nervous system lymphomas using circulating tumor DNA
State-of-the-art response assessment of central nervous system lymphoma (CNSL) by magnetic resonance imaging is challenging and an insufficient predictor of treatment outcomes. Accordingly, the development of novel risk stratification strategies in CNSL is a high unmet medical need. We applied ultrasensitive circulating tumor DNA (ctDNA) sequencing to 146 plasma and cerebrospinal fluid (CSF) samples from 67 patients, aiming to develop an entirely noninvasive dynamic risk model considering clinical and molecular features of CNSL. Our ultrasensitive method allowed for the detection of CNSL-derived mutations in plasma ctDNA with high concordance to CSF and tumor tissue. Undetectable plasma ctDNA at baseline was associated with favorable outcomes. We tracked tumor-specific mutations in plasma-derived ctDNA over time and developed a novel CNSL biomarker based on this information: peripheral residual disease (PRD). Persistence of PRD after treatment was highly predictive of relapse. Integrating established baseline clinical risk factors with assessment of radiographic response and PRD during treatment resulted in the development and independent validation of a novel tool for risk stratification: molecular prognostic index for CNSL (MOP-C). MOP-C proved to be highly predictive of outcomes in patients with CNSL (failure-free survival hazard ratio per risk group of 6.60; 95% confidence interval, 3.12-13.97; P < .0001) and is publicly available at www.mop-c.com. Our results highlight the role of ctDNA sequencing in CNSL. MOP-C has the potential to improve the current standard of clinical risk stratification and radiographic response assessment in patients with CNSL, ultimately paving the way toward individualized treatment.
-
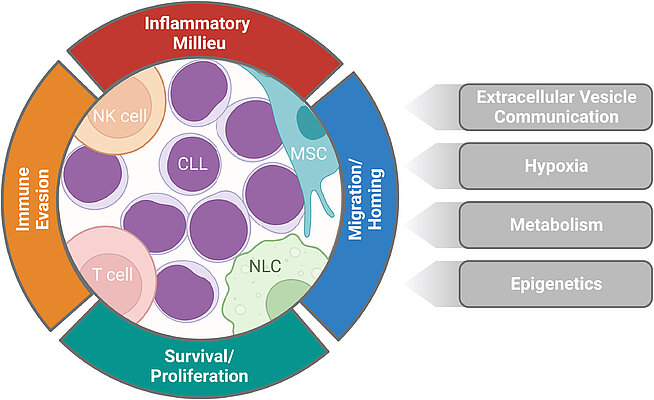
Role of the tumor microenvironment in CLL pathogenesis
Chronic lymphocytic leukemia (CLL) cells extensively interact with and depend on their surrounding tumor microenvironment (TME). The TME encompasses a heterogeneous array of cell types, soluble signals, and extracellular vesicles, which contribute significantly to CLL pathogenesis. CLL cells and the TME cooperatively generate a chronic inflammatory milieu, which reciprocally reprograms the TME and activates a signaling network within CLL cells, promoting their survival and proliferation. Additionally, the inflammatory milieu exerts chemotactic effects, attracting CLL cells and other immune cells to the lymphoid tissues. The intricate CLL-TME interactions also facilitate immune evasion and compromise leukemic cell surveillance. We also review recent advances that have shed light on additional aspects that are substantially influenced by the CLL-TME interplay.
-
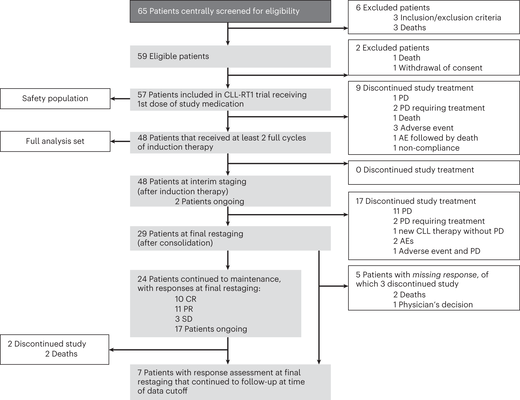
Tislelizumab plus zanubrutinib for Richter transformation: the phase 2 RT1 trial
In patients with chronic lymphocytic leukemia, Richter transformation (RT) reflects the development of an aggressive lymphoma that is associated with poor response to chemotherapy and short survival. We initiated an international, investigator-initiated, prospective, open-label phase 2 study in which patients with RT received a combination of the PD-1 inhibitor tislelizumab plus the BTK inhibitor zanubrutinib for 12 cycles. Patients responding to treatment underwent maintenance treatment with both agents. The primary end point was overall response rate after six cycles. Of 59 enrolled patients, 48 patients received at least two cycles of treatment and comprised the analysis population according to the study protocol. The median observation time was 13.9 months, the median age was 67 (range 45–82) years. Ten patients (20.8%) had received previous RT-directed therapy. In total, 28 out of 48 patients responded to induction therapy with an overall response rate of 58.3% (95% confidence interval (CI) 43.2–72.4), including 9 (18.8%) complete reponse and 19 (39.6%) partial response, meeting the study’s primary end point by rejecting the predefined null hypothesis of 40% (P = 0.008). Secondary end points included duration of response, progression-free survival and overall survival. The median duration of response was not reached, the median progression-free survival was 10.0 months (95% CI 3.8–16.3). Median overall survival was not reached with a 12-month overall survival rate of 74.7% (95% CI 58.4–91.0). The most common adverse events were infections (18.0%), gastrointestinal disorders (13.0%) and hematological toxicities (11.4%). These data suggest that combined checkpoint and BTK inhibition by tislelizumab plus zanubrutinib is an effective and well-tolerated treatment strategy for patients with RT.
-
An inducible Cd79b mutation confers ibrutinib sensitivity in mouse models of Myd88-driven diffuse large B-cell lymphoma
- In mouse models of Myd88-driven DLBCL, the presence of a Cd79b ITAM mutation results in increased BCR proximal signaling. - The increased BCR signaling activity in Cd79b-mutated models confers a selective ibrutinib sensitivity. Diffuse large B cell lymphoma (DLBCL) is the most common aggressive lymphoma and constitutes a highly heterogenous disease. Recent comprehensive genomic profiling revealed the identity of numerous molecularly-defined DLBCL subtypes, including a cluster which is characterized by recurrent aberrations in MYD88, CD79B and BCL2, as well as various lesions promoting a block in plasma cell differentiation, including PRDM1, TBL1XR1 and SPIB. Here, we generated a series of autochthonous mouse models to mimic this DLBCL cluster and specifically focused on the impact of Cd79b mutations in this setting. We show that canonical Cd79b ITAM mutations do not accelerate Myd88- and BCL2-driven lymphomagenesis. Cd79b-mutant murine DLBCL were enriched for IgM surface expression, reminiscent of their human counterparts. Moreover, Cd79b-mutant lymphomas displayed a robust formation of cytoplasmic signaling complexes involving MYD88, CD79B, MALT1 and BTK. These complexes were disrupted upon pharmacological BTK inhibition. The BTK inhibitor-mediated disruption of these signaling complexes translated into a selective ibrutinib sensitivity of lymphomas harboring combined Cd79b and Myd88 mutations. Altogether, this in-depth cross-species comparison provides a framework for the development of molecularly targeted therapeutic intervention strategies in DLBCL.
-
Mouse models of diffuse large B cell lymphoma
Diffuse large B cell lymphoma (DLBCL) is a genetically highly heterogeneous disease. Yet, to date, the vast majority of patients receive standardized frontline chemo-immune-therapy consisting of an anthracycline backbone. Using these regimens, approximately 65% of patients can be cured, whereas the remaining 35% of patients will face relapsed or refractory disease, which, even in the era of CAR-T cells, is difficult to treat. To systematically tackle this high medical need, it is important to design, generate and deploy suitable in vivo model systems that capture disease biology, heterogeneity and drug response. Recently published, large comprehensive genomic characterization studies, which defined molecular sub-groups of DLBCL, provide an ideal framework for the generation of autochthonous mouse models, as well as an ideal benchmark for cell line-derived or patient-derived mouse models of DLBCL. Here we discuss the current state of the art in the field of mouse modelling of human DLBCL, with a particular focus on disease biology and genetically defined molecular vulnerabilities, as well as potential targeting strategies.
-
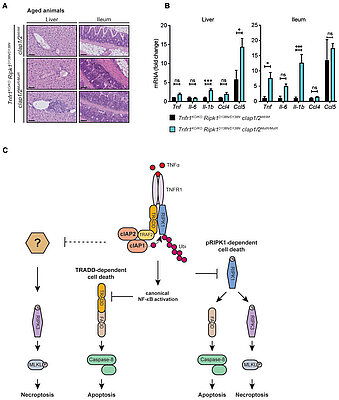
cIAPs control RIPK1 kinase activity-dependent and -independent cell death and tissue inflammation
Cellular inhibitor of apoptosis proteins (cIAPs) are RING-containing E3 ubiquitin ligases that ubiquitylate receptor-interacting protein kinase 1 (RIPK1) to regulate TNF signalling. Here, we established mice simultaneously expressing enzymatically inactive cIAP1/2 variants, bearing mutations in the RING domains of cIAP1/2 (cIAP1/2 mutant RING, cIAP1/2MutR). cIap1/2MutR/MutR mice died during embryonic development due to RIPK1-mediated apoptosis. While expression of kinase-inactive RIPK1D138N rescued embryonic development, Ripk1D138N/D138N/cIap1/2MutR/MutR mice developed systemic inflammation and died postweaning. Cells expressing cIAP1/2MutR and RIPK1D138N were still susceptible to TNF-induced apoptosis and necroptosis, implying additional kinase-independent RIPK1 activities in regulating TNF signalling. Although further ablation of Ripk3 did not lead to any phenotypic improvement, Tnfr1 gene knock-out prevented early onset of systemic inflammation and premature mortality, indicating that cIAPs control TNFR1-mediated toxicity independent of RIPK1 and RIPK3. Beyond providing novel molecular insights into TNF-signalling, the mouse model established in this study can serve as a useful tool to further evaluate ongoing therapeutic protocols using inhibitors of TNF, cIAPs and RIPK1.
-
Cleavage of cFLIP restrains cell death during viral infection and tissue injury and favors tissue repair
Cell death coordinates repair programs following pathogen attack and tissue injury. However, aberrant cell death can interfere with such programs and cause organ failure. Cellular FLICE-like inhibitory protein (cFLIP) is a crucial regulator of cell death and a substrate of Caspase-8. However, the physiological role of cFLIP cleavage by Caspase-8 remains elusive. Here, we found an essential role for cFLIP cleavage in restraining cell death in different pathophysiological scenarios. Mice expressing a cleavage-resistant cFLIP mutant, CflipD377A, exhibited increased sensitivity to severe acute respiratory syndrome coronavirus (SARS-CoV)–induced lethality, impaired skin wound healing, and increased tissue damage caused by Sharpin deficiency. In vitro, abrogation of cFLIP cleavage sensitizes cells to tumor necrosis factor(TNF)–induced necroptosis and apoptosis by favoring complex-II formation. Mechanistically, the cell death–sensitizing effect of the D377A mutation depends on glutamine-469. These results reveal a crucial role for cFLIP cleavage in controlling the amplitude of cell death responses occurring upon tissue stress to ensure the execution of repair programs.
-
Preprint: Pentose Phosphate Pathway Inhibition activates Macrophages towards phagocytic Lymphoma Cell Clearance
Macrophages in the B-cell lymphoma microenvironment represent a functional node in progression and therapeutic response. We assessed metabolic regulation of macrophages in the context of therapeutic antibody-mediated phagocytosis. Pentose phosphate pathway (PPP) inhibition by specific compounds and shRNA targeting induced increased phagocytic lymphoma cell clearance. Moreover, macrophages provided decreased support for survival of lymphoma cells. PPP inhibition induced metabolic activation, cytoskeletal re-modelling and pro-inflammatory polarization of macrophages. A link between PPP and immune regulation was identified as mechanism of macrophage repolarization. Inhibition of the PPP causes suppression of glycogen synthesis and subsequent modulation of the immune modulatory UDPG-Stat1-Irg1-Itaconate axis. PPP inhibition rewired macrophage maturation and activation in vivo. Addition of the PPP inhibitor S3 to antibody therapy achieved significantly prolonged overall survival in an aggressive B-cell lymphoma mouse model. We hypothesize the PPP as key regulator and targetable modulator of macrophage activity in lymphoma to improve efficacy of immunotherapies. - Macrophage-mediated lymphoma cell phagocytosis is increased by pentose phosphate pathway (PPP) inhibition as an immune regulatory switch for macrophage function and polarization - PPP inhibition is linked to decreased glycogen synthesis and subsequent modulation of the UDPG-Stat1-Irg1-Itaconate axis - PPP inhibition is tolerable in vivo and facilitates therapeutic targeting of B-cell lymphoma
-
First-Line Venetoclax Combinations in Chronic Lymphocytic Leukemia
BACKGROUND Randomized trials of venetoclax plus anti-CD20 antibodies as first-line treatment in fit patients (i.e., those with a low burden of coexisting conditions) with advanced chronic lymphocytic leukemia (CLL) have been lacking. METHODS In a phase 3, open-label trial, we randomly assigned, in a 1:1:1:1 ratio, fit patients with CLL who did not have TP53 aberrations to receive six cycles of chemoimmunotherapy (fludarabine–cyclophosphamide–rituximab or bendamustine–rituximab) or 12 cycles of venetoclax–rituximab, venetoclax–obinutuzumab, or venetoclax–obinutuzumab–ibrutinib. Ibrutinib was discontinued after two consecutive measurements of undetectable minimal residual disease or could be extended. The primary end points were undetectable minimal residual disease (sensitivity, <10−4 [i.e., <1 CLL cell in 10,000 leukocytes]) as assessed by flow cytometry in peripheral blood at month 15 and progression-free survival. [...] CONCLUSIONS Venetoclax–obinutuzumab with or without ibrutinib was superior to chemoimmunotherapy as first-line treatment in fit patients with CLL. (Funded by AbbVie and others; GAIA–CLL13 ClinicalTrials.gov number, NCT02950051.; EudraCT number, 2015-004936-36.)
-
Transcriptomic profiles and 5-year results from the randomized CLL14 study of venetoclax plus obinutuzumab versus chlorambucil plus obinutuzumab in chronic lymphocytic leukemia
Data on long-term outcomes and biological drivers associated with depth of remission after BCL2 inhibition by venetoclax in the treatment of chronic lymphocytic leukemia (CLL) are limited. In this open-label parallel-group phase-3 study, 432 patients with previously untreated CLL were randomized (1:1) to receive either 1-year venetoclax-obinutuzumab (Ven-Obi, 216 patients) or chlorambucil-Obi (Clb-Obi, 216 patients) therapy (NCT02242942). The primary endpoint was investigator-assessed progression-free survival (PFS); secondary endpoints included minimal residual disease (MRD) and overall survival. RNA sequencing of CD19-enriched blood was conducted for exploratory post-hoc analyses. After a median follow-up of 65.4 months, PFS is significantly superior for Ven-Obi compared to Clb-Obi (Hazard ratio [HR] 0.35 [95% CI 0.26–0.46], p < 0.0001). At 5 years after randomization, the estimated PFS rate is 62.6% after Ven-Obi and 27.0% after Clb-Obi. In both arms, MRD status at the end of therapy is associated with longer PFS. MRD + ( ≥ 10−4) status is associated with increased expression of multi-drug resistance gene ABCB1 (MDR1), whereas MRD6 (< 10−6) is associated with BCL2L11 (BIM) expression. Inflammatory response pathways are enriched in MRD+ patient solely in the Ven-Obi arm. These data indicate sustained long-term efficacy of fixed-duration Ven-Obi in patients with previously untreated CLL. The distinct transcriptomic profile of MRD+ status suggests possible biological vulnerabilities.
-
Non-viral TRAC-knocked-in CD19KICAR-T and gp350KICAR-T cells tested against Burkitt lymphomas with type 1 or 2 EBV infection: In vivo cellular dynamics and potency
Introduction: The ubiquitous Epstein–Barr virus (EBV) is an oncogenic herpes virus associated with several human malignancies. EBV is an immune-evasive pathogen that promotes CD8+ T cell exhaustion and dysregulates CD4+ T cell functions. Burkitt lymphoma (BL) is frequently associated with EBV infections. Since BL relapses after conventional therapies are difficult to treat, we evaluated prospective off-the-shelf edited CAR-T cell therapies targeting CD19 or the EBV gp350 cell surface antigen. Methods: We used CRISPR/Cas9 gene editing methods to knock in (KI) the CD19CAR.CD28z or gp350CAR.CD28z into the T cell receptor (TCR) alpha chain (TRAC) locus.
-
Mitochondrial pores at the crossroad between cell death and inflammatory signaling
Mitochondria are cellular organelles with a major role in many cellular processes, including not only energy production, metabolism, and calcium homeostasis but also regulated cell death and innate immunity. Their proteobacterial origin makes them a rich source of potent immune agonists, normally hidden within the mitochondrial membrane barriers. Alteration of mitochondrial permeability through mitochondrial pores thus provides efficient mechanisms not only to communicate mitochondrial stress to the cell but also as a key event in the integration of cellular responses. In this regard, eukaryotic cells have developed diverse signaling networks that sense and respond to the release of mitochondrial components into the cytosol and play a key role in controlling cell death and inflammatory pathways. Modulating pore formation at mitochondria through direct or indirect mechanisms may thus open new opportunities for therapy. In this review, we discuss the current understanding of the structure and molecular mechanisms of mitochondrial pores and how they function at the interface between cell death and inflammatory signaling to regulate cellular outcomes.
-
LYN kinase programs stromal fibroblasts to facilitate leukemic survival via regulation of c-JUN and THBS1
Microenvironmental bystander cells are essential for the progression of chronic lymphocytic leukemia (CLL). We have discovered previously that LYN kinase promotes the formation of a microenvironmental niche for CLL. Here we provide mechanistic evidence that LYN regulates the polarization of stromal fibroblasts to support leukemic progression. LYN is overexpressed in fibroblasts of lymph nodes of CLL patients. LYN-deficient stromal cells reduce CLL growth in vivo. LYN-deficient fibroblasts show markedly reduced leukemia feeding capacity in vitro. Multi-omics profiling reveals that LYN regulates the polarization of fibroblasts towards an inflammatory cancer-associated phenotype through modulation of cytokine secretion and extracellular matrix composition. Mechanistically, LYN deletion reduces inflammatory signaling including reduction of c-JUN expression, which in turn augments the expression of Thrombospondin-1, which binds to CD47 thereby impairing CLL viability. Together, our findings suggest that LYN is essential for rewiring fibroblasts towards a leukemia-supportive phenotype.
-
Focal structural variants revealed by whole genome sequencing disrupt the histone demethylase KDM4C in B-cell lymphomas
Histone methylation-modifiers, such as EZH2 and KMT2D, are recurrently altered in B-cell lymphomas. To comprehensively describe the landscape of alterations affecting genes encoding histone methylation-modifiers in lymphomagenesis we investigated whole genome and transcriptome data of 186 mature B-cell lymphomas sequenced in the ICGC MMML-Seq project. Besides confirming common alterations of KMT2D (47% of cases), EZH2 (17%), SETD1B (5%), PRDM9 (4%), KMT2C (4%), and SETD2 (4%), also identified by prior exome or RNA-sequencing studies, we here found recurrent alterations to KDM4C in chromosome 9p24, encoding a histone demethylase. Focal structural variation was the main mechanism of KDM4C alterations, and was independent from 9p24 amplification. We also identified KDM4C alterations in lymphoma cell lines including a focal homozygous deletion in a classical Hodgkin lymphoma cell line. By integrating RNA-sequencing and genome sequencing data we predict that KDM4C structural variants result in loss-offunction. By functional reconstitution studies in cell lines, we provide evidence that KDM4C can act as a tumor suppressor. Thus, we show that identification of structural variants in whole genome sequencing data adds to the comprehensive description of the mutational landscape of lymphomas and, moreover, establish KDM4C as a putative tumor suppressive gene recurrently altered in subsets of B-cell derived lymphomas.
-
Distinct Genetically Determined Origins of Myd88/BCL2-Driven Aggressive Lymphoma Rationalize Targeted Therapeutic Intervention Strategies
Genomic profiling revealed the identity of at least 5 subtypes of diffuse large B-cell lymphoma (DLBCL), including the MCD/C5 cluster characterized by aberrations in MYD88, BCL2, PRDM1, and/or SPIB. We generated mouse models harboring B cell–specific Prdm1 or Spib aberrations on the background of oncogenic Myd88 and Bcl2 lesions. We deployed whole-exome sequencing, transcriptome, flow-cytometry, and mass cytometry analyses to demonstrate that Prdm1- or Spib-altered lymphomas display molecular features consistent with prememory B cells and light-zone B cells, whereas lymphomas lacking these alterations were enriched for late light-zone and plasmablast-associated gene sets. Consistent with the phenotypic evidence for increased B cell receptor signaling activity in Prdm1-altered lymphomas, we demonstrate that combined BTK/BCL2 inhibition displays therapeutic activity in mice and in five of six relapsed/refractory DLBCL patients. Moreover, Prdm1-altered lymphomas were immunogenic upon transplantation into immuno-competent hosts, displayed an actionable PD-L1 surface expression, and were sensitive to antimurine-CD19-CAR-T cell therapy, in vivo.
-
Zanubrutinib or Ibrutinib in Relapsed or Refractory Chronic Lymphocytic Leukemia
Background: In a multinational, phase 3, head-to-head trial, ibrutinib, a Bruton's tyrosine kinase (BTK) inhibitor, was compared with zanubrutinib, a BTK inhibitor with greater specificity, as treatment for relapsed or refractory chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL). In prespecified interim analyses, zanubrutinib was superior to ibrutinib with respect to overall response (the primary end point). Data from the final analysis of progression-free survival are now available. In patients with relapsed or refractory CLL or SLL, progression-free survival was significantly longer among patients who received zanubrutinib than among those who received ibrutinib, and zanubrutinib was associated with fewer cardiac adverse events. (Funded by BeiGene; ALPINE ClinicalTrials.gov number, NCT03734016.).
-
An Aged/Autoimmune B-cell Program Defines the Early Transformation of Extranodal Lymphomas
A third of patients with diffuse large B-cell lymphoma (DLBCL) present with extranodal dissemination, which is associated with inferior clinical outcomes. MYD88L265P is a hallmark extranodal DLBCL mutation that supports lymphoma proliferation. Yet extranodal lymphomagenesis and the role of MYD88L265P in transformation remain mostly unknown. Here, we show that B cells expressing Myd88L252P (MYD88L265P murine equivalent) activate, proliferate, and differentiate with minimal T-cell costimulation. Additionally, Myd88L252P skewed B cells toward memory fate. Unexpectedly, the transcriptional and phenotypic profiles of B cells expressing Myd88L252P, or other extranodal lymphoma founder mutations, resembled those of CD11c+T-BET+ aged/autoimmune memory B cells (AiBC). AiBC-like cells progressively accumulated in animals prone to develop lymphomas, and ablation of T-BET, the AiBC master regulator, stripped mouse and human mutant B cells of their competitive fitness. By identifying a phenotypically defined prospective lymphoma precursor population and its dependencies, our findings pave the way for the early detection of premalignant states and targeted prophylactic interventions in high-risk patients.
-
Outcome Prediction in Patients With Large B-cell Lymphoma Undergoing Chimeric Antigen Receptor T-cell Therapy
The introduction of chimeric antigen receptor (CAR) T-cell therapy has led to a fundamental shift in the management of relapsed and refractory large B-cell lymphoma. However, our understanding of risk factors associated with non-response is still insufficient and the search for predictive biomarkers continues. Some parameters measurable on 18F-fluorodeoxyglucose positron emission tomography (PET) may be of additional value in this context. A total of 47 individuals from three German university centers who underwent re-staging with PET prior to CAR T-cell therapy were enrolled into the present study. After multivariable analysis considering tumor characteristics and patient factors that might affect progression-free survival (PFS), we investigated whether metabolic tumor volume (MTV) or maximum standardized uptake value (SUVmax) further improve risk stratification. Their most suitable cut-offs were determined by Cox and logistic regression. Forward selection identified extra-nodal disease as the most predictive factor of those routinely available, and we found it to be associated with significantly inferior overall survival after CAR T-cell treatment (P = 0.012). Furthermore, patients with MTV and SUVmax higher than the optimal threshold of 11 mL and 16.7, respectively, experienced shorter PFS (P = 0.016 and 0.002, respectively). Hence, these risk factors might be useful for selection of individuals likely to benefit from CAR T-cell therapy and their management.
-
BCL-2-family protein tBID can act as a BAX-like effector of apoptosis
During apoptosis, the BCL-2-family protein tBID promotes mitochondrial permeabilization by activating BAX and BAK and by blocking anti-apoptotic BCL-2 members. Here, we report that tBID can also mediate mitochondrial permeabilization by itself, resulting in release of cytochrome c and mitochondrial DNA, caspase activation and apoptosis even in absence of BAX and BAK. This previously unrecognized activity of tBID depends on helix 6, homologous to the pore-forming regions of BAX and BAK, and can be blocked by pro-survival BCL-2 proteins. Importantly, tBID-mediated mitochondrial permeabilization independent of BAX and BAK is physiologically relevant for SMAC release in the immune response against Shigella infection. Furthermore, it can be exploited to kill leukaemia cells with acquired venetoclax resistance due to lack of active BAX and BAK. Our findings define tBID as an effector of mitochondrial permeabilization in apoptosis and provide a new paradigm for BCL-2 proteins, with implications for anti-bacterial immunity and cancer therapy.
-
Deregulation and epigenetic modification of BCL2-family genes cause resistance to venetoclax in hematologic malignancies
The BCL2 inhibitor venetoclax has been approved to treat different hematological malignancies. Because there is no common genetic alteration causing resistance to venetoclax in chronic lymphocytic leukemia (CLL) and B-cell lymphoma, we asked if epigenetic events might be involved in venetoclax resistance. Therefore, we employed whole-exome sequencing, methylated DNA immunoprecipitation sequencing, and genome-wide clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 screening to investigate venetoclax resistance in aggressive lymphoma and high-risk CLL patients. We identified a regulatory CpG island within the PUMA promoter that is methylated upon venetoclax treatment, mediating PUMA downregulation on transcript and protein level. PUMA expression and sensitivity toward venetoclax can be restored by inhibition of methyltransferases. We can demonstrate that loss of PUMA results in metabolic reprogramming with higher oxidative phosphorylation and adenosine triphosphate production, resembling the metabolic phenotype that is seen upon venetoclax resistance. Although PUMA loss is specific for acquired venetoclax resistance but not for acquired MCL1 resistance and is not seen in CLL patients after chemotherapy-resistance, BAX is essential for sensitivity toward both venetoclax and MCL1 inhibition. As we found loss of BAX in Richter's syndrome patients after venetoclax failure, we defined BAX-mediated apoptosis to be critical for drug resistance but not for disease progression of CLL into aggressive diffuse large B-cell lymphoma in vivo. A compound screen revealed TRAIL-mediated apoptosis as a target to overcome BAX deficiency. Furthermore, antibody or CAR T cells eliminated venetoclax resistant lymphoma cells, paving a clinically applicable way to overcome venetoclax resistance.
-
Interim FDG-PET analysis to identify patients with aggressive non-Hodgkin lymphoma who benefit from treatment intensification: a post-hoc analysis of the PETAL trial
The randomized PETAL trial failed to demonstrate a benefit of interim FDG-PET (iPET)-based treatment intensification over continued standard therapy with CHOP (plus rituximab (R) in CD20-positive lymphomas). We hypothesized that PET analysis of all lymphoma manifestations may identify patients who benefitted from treatment intensification. A previously developed neural network was employed for iPET analysis to identify the highest pathological FDG uptake (max-SUVAI) and the mean FDG uptake of all lymphoma manifestations (mean-SUVAI). High mean-SUVAI uptake was determined separately for iPET-positive and iPET-negative patients. The endpoint was time-to-progression (TTP). There was a significant interaction of additional rituximab and mean-SUVAI in the iPET-negative group (HR = 0.6, p < 0.05). Patients with high mean-SUVAI had significantly prolonged TTP when treated with 6xR-CHOP + 2 R (not reached versus 52 months, p < 0.05), whereas max-SUVmanual failed to show an impact of additional rituximab. In the iPET-positive group, patients with high mean-SUVAI had a significantly longer TTP with (R-)CHOP than with the Burkitt protocol (14 versus 4 months, p < 0.01). Comprehensive iPET evaluation may provide new prognosticators in aggressive lymphoma. Additional application of rituximab was associated with prolonged TTP in iPET-negative patients with high mean-SUVAI. Comprehensive iPET interpretation could identify high-risk patients who benefit from study-specific interventions.
-
Tripartite antigen-agnostic combination immunotherapy cures established poorly immunogenic tumors
Single-agent immunotherapy has shown remarkable efficacy in selected cancer entities and individual patients. However, most patients fail to respond. This is likely due to diverse immunosuppressive mechanisms acting in a concerted way to suppress the host anti-tumor immune response. Combination immunotherapy approaches that are effective in such poorly immunogenic tumors mostly rely on precise knowledge of antigenic determinants on tumor cells. Creating an antigen-agnostic combination immunotherapy that is effective in poorly immunogenic tumors for which an antigenic determinant is not known is a major challenge.
-
MLKL post-translational modifications: road signs to infection, inflammation and unknown destinations
Necroptosis is a caspase-independent modality of cell death that requires the activation of the executioner MLKL. In the last ten years the field gained a substantial amount of evidence regarding its involvement in host response to pathogens, TNF-induced inflammatory diseases as well as pathogen recognition receptors (PRR)-induced inflammation. However, there are still a lot of questions that remain unanswered. While it is clear that there are specific events needed to drive MLKL activation, substantial differences between human and mouse MLKL not only highlight different evolutionary pressure, but also provide potential insights on alternative modalities of activation. While in TNF-induced necroptosis it is clear the involvement of the RIPK3 mediated phosphorylation, it still remains to be understood how certain inflammatory in vivo phenotypes are not equally rescued by either RIPK3 or MLKL loss. Moreover, the plethora of different reported phosphorylation events on MLKL, even in cells that do not express RIPK3, suggest indeed that there is more to MLKL than RIPK3-mediated activation, not only in the execution of necroptosis but perhaps in other inflammatory conditions that include IFN response. The recent discovery of MLKL ubiquitination has highlighted a new checkpoint in the regulation of MLKL activation and the somewhat conflicting evidence reported certainly require some untangling. In this review we will highlight the recent findings on MLKL activation and involvement to pathogen response with a specific focus on MLKL post-translational modifications, in particular ubiquitination. This review will highlight the outstanding main questions that have risen from the last ten years of research, trying at the same time to propose potential avenues of research.
-
Aberrant expansion of spontaneous splenic germinal centers induced by hallmark genetic lesions of aggressive lymphoma
Unique molecular vulnerabilities have been identified in the aggressive MCD/C5 genetic subclass of diffuse large B-cell lymphoma (DLBCL). However, the premalignant cell-of-origin exhibiting MCD-like dependencies remains elusive. In this study, we examined animals carrying up to 4 hallmark genetic lesions found in MCD consisting of gain-of-function mutations in Myd88 and Cd79b, loss of Prdm1, and overexpression of BCL2. We discovered that expression of combinations of these alleles in vivo promoted a cell-intrinsic accumulation of B cells in spontaneous splenic germinal centers (GCs). As with MCD, these premalignant B cells were enriched for B-cell receptors (BCRs) with evidence of self-reactivity, displayed a de novo dependence on Tlr9, and were more sensitive to inhibition of Bruton's tyrosine kinase. Mutant spontaneous splenic GC B cells (GCB) showed increased proliferation and IRF4 expression. Mice carrying all 4 genetic lesions showed a >50-fold expansion of spontaneous splenic GCs exhibiting aberrant histologic features with a dark zone immunophenotype and went on to develop DLBCL in the spleen with age. Thus, by combining multiple hallmark genetic alterations associated with MCD, our study identifies aberrant spontaneous splenic GCBs as a likely cell-of-origin for this aggressive genetic subtype of lymphoma.
-
Concomitant Cytotoxic Effector Differentiation of CD4+ and CD8+ T Cells in Response to EBV-Infected B Cells
Most people infected by EBV acquire specific immunity, which then controls latent infection throughout their life. Immune surveillance of EBV-infected cells by cytotoxic CD4+ T cells has been recognized; however, the molecular mechanism of generating cytotoxic effector T cells of the CD4+ subset remains poorly understood. Here we compared phenotypic features and the transcriptome of EBV-specific effector-memory CD4+ T cells and CD8+ T cells in mice and found that both T cell types show cytotoxicity and, to our surprise, widely similar gene expression patterns relating to cytotoxicity. Similar to cytotoxic CD8+ T cells, EBV-specific cytotoxic CD4+ T cells from human peripheral blood expressed T-bet, Granzyme B, and Perforin and upregulated the degranulation marker, CD107a, immediately after restimulation. Furthermore, T-bet expression in cytotoxic CD4+ T cells was highly correlated with Granzyme B and Perforin expression at the protein level. Thus, differentiation of EBV-specific cytotoxic CD4+ T cells is possibly controlled by mechanisms shared by cytotoxic CD8+ T cells. T-bet-mediated transcriptional regulation may explain the similarity of cytotoxic effector differentiation between CD4+ T cells and CD8+ T cells, implicating that this differentiation pathway may be directed by environmental input rather than T cell subset.
-
Regulatory CAR-T cells in autoimmune diseases: Progress and current challenges
CAR (Chimeric Antigen Receptor) T-cell therapy has revolutionized the field of oncology in recent years. This innovative shift in cancer treatment also provides the opportunity to improve therapies for many patients suffering from various autoimmune diseases. Recent studies have confirmed the therapeutic suppressive potential of regulatory T cells (Tregs) to modulate immune response in autoimmune diseases. However, the polyclonal character of regulatory T cells and their unknown TCR specificity impaired their therapeutic potency in clinical implementation. Genetical engineering of these immune modulating cells to express antigen-specific receptors and using them therapeutically is a logical step on the way to overcome present limitations of the Treg strategy for the treatment of autoimmune diseases. Encouraging preclinical studies successfully demonstrated immune modulating properties of CAR Tregs in various mouse models. Still, there are many concerns about targeted Treg therapies relating to CAR target selectivity, suppressive functions, phenotype stability and safety aspects. Here, we summarize recent developments in CAR design, Treg biology and future strategies and perspectives in CAR Treg immunotherapy aiming at clinical translation.
-
Extracellular vesicles and PD-L1 suppress macrophages, inducing therapy resistance in TP53-deficient B-cell malignancies
Genetic alterations in the DNA damage response (DDR) pathway are a frequent mechanism of resistance to chemoimmunotherapy (CIT) in B-cell malignancies. We have previously shown that the synergy of CIT relies on secretory crosstalk elicited by chemotherapy between the tumor cells and macrophages. Here, we show that loss of multiple different members of the DDR pathway inhibits macrophage phagocytic capacity in vitro and in vivo. Particularly, loss of TP53 led to decreased phagocytic capacity ex vivo across multiple B-cell malignancies. We demonstrate via in vivo cyclophosphamide treatment using the Eμ-TCL1 mouse model that loss of macrophage phagocytic capacity in Tp53-deleted leukemia is driven by a significant downregulation of a phagocytic transcriptomic signature using small conditional RNA sequencing. By analyzing the tumor B-cell proteome, we identified a TP53-specific upregulation of proteins associated with extracellular vesicles (EVs). We abrogated EV biogenesis in tumor B-cells via clustered regularly interspaced short palindromic repeats (CRISPR)-knockout (KO) of RAB27A and confirmed that the EVs from TP53-deleted lymphoma cells were responsible for the reduced phagocytic capacity and the in vivo CIT resistance. Furthermore, we observed that TP53 loss led to an upregulation of both PD-L1 cell surface expression and secretion of EVs by lymphoma cells. Disruption of EV bound PD-L1 by anti-PD-L1 antibodies or PD-L1 CRISPR-KO improved macrophage phagocytic capacity and in vivo therapy response. Thus, we demonstrate enhanced EV release and increased PD-L1 expression in TP53-deficient B-cell lymphomas as novel mechanisms of macrophage function alteration in CIT resistance. This study indicates the use of checkpoint inhibition in the combination treatment of B-cell malignancies with TP53 loss.
-
Sustainable Clinical Development of CAR-T Cells – Switching From Viral Transduction Towards CRISPR-Cas Gene Editing
T cells modified for expression of Chimeric Antigen Receptors (CARs) were the first gene-modified cell products approved for use in cancer immunotherapy. CAR-T cells engineered with gammaretroviral or lentiviral vectors (RVs/LVs) targeting B-cell lymphomas and leukemias have shown excellent clinical efficacy and no malignant transformation due to insertional mutagenesis to date. Large-scale production of RVs/LVs under good-manufacturing practices for CAR-T cell manufacturing has soared in recent years. However, manufacturing of RVs/LVs remains complex and costly, representing a logistical bottleneck for CAR-T cell production. Emerging gene-editing technologies are fostering a new paradigm in synthetic biology for the engineering and production of CAR-T cells. Firstly, the generation of the modular reagents utilized for gene editing with the CRISPR-Cas systems can be scaled-up with high precision under good manufacturing practices, are interchangeable and can be more sustainable in the long-run through the lower material costs. Secondly, gene editing exploits the precise insertion of CARs into defined genomic loci and allows combinatorial gene knock-ins and knock-outs with exciting and dynamic perspectives for T cell engineering to improve their therapeutic efficacy. Thirdly, allogeneic edited CAR-effector cells could eventually become available as "off-the-shelf" products. This review addresses important points to consider regarding the status quo, pending needs and perspectives for the forthright evolution from the viral towards gene editing developments for CAR-T cells.
-
Efficacy and Safety of Tirabrutinib and Idelalisib With or Without Obinutuzumab in Relapsed Chronic Lymphocytic Leukemia
-
Cell Death-Related Ubiquitin Modifications in Inflammatory Syndromes: From Mice to Men
Aberrant cell death can cause inflammation and inflammation-related diseases. While the link between cell death and inflammation has been widely established in mouse models, evidence supporting a role for cell death in the onset of inflammatory and autoimmune diseases in patients is still missing. In this review, we discuss how the lessons learnt from mouse models can help shed new light on the initiating or contributing events leading to immune-mediated disorders. In addition, we discuss how multiomic approaches can provide new insight on the soluble factors released by dying cells that might contribute to the development of such diseases.
-
The scaffold protein NEDD9 is necessary for leukemia-cell migration and disease progression in a mouse model of chronic lymphocytic leukemia
The scaffold protein NEDD9 is frequently upregulated and hyperphosphorylated in cancers, and is associated with poor clinical outcome. NEDD9 promotes B-cell adhesion, migration and chemotaxis, pivotal processes for malignant development. We show that global or B-cell-specific deletion of Nedd9 in chronic lymphocytic leukemia (CLL) mouse models delayed CLL development, markedly reduced disease burden and resulted in significant survival benefit. NEDD9 was required for efficient CLL cell homing, chemotaxis, migration and adhesion. In CLL patients, peripheral NEDD9 expression was associated with adhesion and migration signatures as well as leukocyte count. Additionally, CLL lymph nodes frequently expressed high NEDD9 levels, with a subset of patients showing NEDD9 expression enriched in the CLL proliferation centers. Blocking activity of prominent NEDD9 effectors, including AURKA and HDAC6, effectively reduced CLL cell migration and chemotaxis. Collectively, our study provides evidence for a functional role of NEDD9 in CLL pathogenesis that involves intrinsic defects in adhesion, migration and homing.
-
Loss of function mutations of BCOR in classical Hodgkin lymphoma
BCOR is a component of a variant Polycomb repressive complex 1 (PRC1.1). PRC1 and PRC2 complexes together constitute a major gene regulatory system critical for appropriate cellular differentiation. The gene is upregulated in germinal center (GC) B cells and mutated in a number of hematologic malignancies. We report BCOR inactivating alterations in 4/7 classic Hodgkin lymphoma (cHL) cell lines, subclonal somatic mutations in Hodgkin and Reed-Sternberg (HRS) cells of 4/10 cHL cases, and deletions in HRS cells of 7/17 primary cHL cases. In mice, conditional loss of Bcor driven by AID-Cre in GC B cells resulted in gene expression changes of 46 genes (>2-fold) including upregulated Lef1 that encodes a transcription factor responsible for establishing T-cell identity and Il9r (interleukin-9 receptor), an important member of the cytokine network in cHL. Our findings suggest a role for BCOR loss in cHL pathogenesis and GC-B cell homeostasis.
-
DRP1 interacts directly with BAX to induce its activation and apoptosis
The apoptotic executioner protein BAX and the dynamin‐like protein DRP1 co‐localize at mitochondria during apoptosis to mediate mitochondrial permeabilization and fragmentation. However, the molecular basis and functional consequences of this interplay remain unknown. Here, we show that BAX and DRP1 physically interact, and that this interaction is enhanced during apoptosis. Complex formation between BAX and DRP1 occurs exclusively in the membrane environment and requires the BAX N‐terminal region, but also involves several other BAX surfaces. Furthermore, the association between BAX and DRP1 enhances the membrane activity of both proteins. Forced dimerization of BAX and DRP1 triggers their activation and translocation to mitochondria, where they induce mitochondrial remodeling and permeabilization to cause apoptosis even in the absence of apoptotic triggers. Based on this, we propose that DRP1 can promote apoptosis by acting as noncanonical direct activator of BAX through physical contacts with its N‐terminal region.
-
Efficacy and Safety of the Combination of Tirabrutinib and Entospletinib With or Without Obinutuzumab in Relapsed Chronic Lymphocytic Leukemia
-
Potent pro-apoptotic combination therapy is highly effective in a broad range of cancers
Primary or acquired therapy resistance is a major obstacle to the effective treatment of cancer. Resistance to apoptosis has long been thought to contribute to therapy resistance. We show here that recombinant TRAIL and CDK9 inhibition cooperate in killing cells derived from a broad range of cancers, importantly without inducing detectable adverse events. Remarkably, the combination of TRAIL with CDK9 inhibition was also highly effective on cancers resistant to both, standard-of-care chemotherapy and various targeted therapeutic approaches. Dynamic BH3 profiling revealed that, mechanistically, combining TRAIL with CDK9 inhibition induced a drastic increase in the mitochondrial priming of cancer cells. Intriguingly, this increase occurred irrespective of whether the cancer cells were sensitive or resistant to chemo- or targeted therapy. We conclude that this pro-apoptotic combination therapy has the potential to serve as a highly effective new treatment option for a variety of different cancers. Notably, this includes cancers that are resistant to currently available treatment modalities.
-
Linear ubiquitin as a common regulator of cellular stress
Linear or M1-ubiquitination (Ub) is required for optimal NF-kB activation and for cell death inhibition. Using Drosophila as a model organism, Aalto et al. found that hypoxia, oxidative and mechanical stress induced M1-Ub by the HOIP homolog, LUBEL. Increased M1-Ub had a protective function driven by activation of the NF-κB transcription factor Relish via the Immune deficiency pathway (Imd). This protective M1-Ub was also induced upon cellular stress in colorectal cancer cells. Collectively, they propose that M1-Ub is a conserved, common response to different forms of stresses. These findings may have important implications for the use of HOIP inhibitors for cancer treatment.
-
The interplay between BAX and BAK tunes apoptotic pore growth to control mitochondrial-DNA-mediated inflammation
BAX and BAK are key apoptosis regulators that mediate the decisive step of mitochondrial outer membrane permeabilization. However, the mechanism by which they assemble the apoptotic pore remains obscure. Here, we report that BAX and BAK present distinct oligomerization properties, with BAK organizing into smaller structures with faster kinetics than BAX. BAK recruits and accelerates BAX assembly into oligomers that continue to grow during apoptosis. As a result, BAX and BAK regulate each other as they co-assemble into the same apoptotic pores, which we visualize. The relative availability of BAX and BAK molecules thereby determines the growth rate of the apoptotic pore and the relative kinetics by which mitochondrial contents, most notably mtDNA, are released. This feature of BAX and BAK results in distinct activation kinetics of the cGAS/STING pathway with implications for mtDNA-mediated paracrine inflammatory signaling.
-
Evaluation of a Prognostic Epigenetic Classification System in Chronic Lymphocytic Leukemia Patients
Methylation at 5 CpG sites was previously shown to classify chronic lymphocytic leukemia (CLL) into 3 prognostic subgroups. Here, we aimed to validate the marker set in an additional cohort and to evaluate its clinical utility for CLL patient stratification. We evaluated this epigenetic marker set in 79 German patients using bisulfite treatment followed by pyrosequencing and classification using a support vector machine-learning tool. The n-CLL, i-CLL, and m-CLL classification was detected in 28 (35%), 10 (13%), and 41 (51%) patients, respectively. Epigenetic grouping was associated with IGHV mutational status (P = 2 × 10-12), isolated del13q (P = 9 × 10-6), del17p (P = .015), complex karyotype (P = .005), VH-usage, and clinical outcome as time to first treatment (P = 1.4 × 10-12) and overall survival (P = .003). Multivariate Cox regression analysis identified n-CLL as a factor for earlier treatment hazard ratio (HR), 6.3 (95% confidence interval [CI] 2.4-16.4; P = .0002) compared to IGHV mutational status (HR 4.6, 95% CI 1.9-11.3, P = .0008). In addition, when comparing the prognostic value of the epigenetic classification system with the IGHV classification, epigenetic grouping performed better compared to IGHV mutational status using Kaplan-Meier estimation and allowed the identification of a third, intermediate (i-CLL) group. Thus, our study confirmed the prognostic value of the epigenetic marker set for patient stratification in routine clinical diagnostics.
-
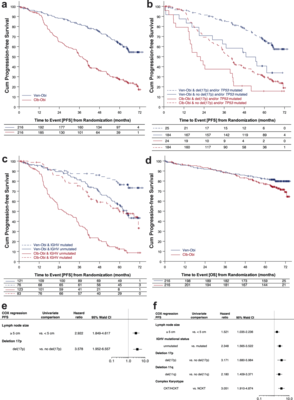
Minimal Residual Disease Dynamics after Venetoclax-Obinutuzumab Treatment: Extended Off-Treatment Follow-up From the Randomized CLL14 Study
The CLL14 study has established one-year fixed-duration treatment of venetoclax and obinutuzumab (Ven-Obi) for patients with previously untreated chronic lymphocytic leukemia. With all patients off treatment for at least three years, we report a detailed analysis of minimal residual disease (MRD) kinetics and long-term outcome of patients treated in the CLL14 study. Patients were randomly assigned to receive six cycles of obinutuzumab with 12 cycles of venetoclax or 12 cycles of chlorambucil (Clb-Obi). Progression-free survival (PFS) was the primary end point. Key secondary end points included rates of undetectable MRD and overall survival. To analyze MRD kinetics, a population-based growth model with nonlinear mixed effects approach was developed. Of 432 patients, 216 were assigned to Ven-Obi and 216 to Clb-Obi. Three months after treatment completion, 40% of patients in the Ven-Obi arm (7% in the Clb-Obi arm) had undetectable MRD levels < 10-6 by next-generation sequencing in peripheral blood. Median MRD doubling time was longer after Ven-Obi than Clb-Obi therapy (median 80 v 69 days). At a median follow-up of 52.4 months, a sustained significant PFS improvement was observed in the Ven-Obi arm compared with Clb-Obi (median not reached v 36.4 months; hazard ratio 0.33; 95% CI, 0.25 to 0.45; P < .0001). The estimated 4-year PFS rate was 74.0% in the Ven-Obi and 35.4% in the Clb-Obi arm. No difference in overall survival was observed (hazard ratio 0.85; 95% CI, 0.54 to 1.35; P = .49). No new safety signals occurred. Appearance of MRD after Ven-Obi is significantly slower than that after Clb-Obi with more effective MRD reduction. These findings translate into a superior long-term efficacy with the majority of Ven-Obi-treated patients remaining in remission.
-
Impact of a faulty germinal center reaction on the pathogenesis of primary diffuse large B cell lymphoma of the central nervous system
Primary lymphoma of the central nervous system (PCNSL, CNS) is a specific diffuse large B cell lymphoma (DLBCL) entity confined to the CNS. Key to its pathogenesis is a failure of B cell differentiation and a lack of appropriate control at differentiation stages before entrance and within the germinal center (GC). Self-/polyreactive B cells rescued from apoptosis by MYD88 and/or CD79B mutations accumulate a high load of somatic mutations in their rearranged immunoglobulin (IG) genes, with ongoing somatic hypermutation (SHM). Furthermore, the targeting of oncogenes by aberrant SHM (e.g., PIM1, PAX5, RHOH, MYC, BTG2, KLHL14, SUSD2), translocations of the IG and BCL6 genes, and genomic instability (e.g., gains of 18q21; losses of 9p21, 8q12, 6q21) occur in these cells in the course of their malignant transformation. Activated Toll-like receptor, B cell receptor (BCR), and NF-κB signaling pathways foster lymphoma cell proliferation. Hence, tumor cells are arrested in a late B cell differentiation stage, corresponding to late GC exit B cells, which are genetically related to IgM+ memory cells. Paradoxically, the GC reaction increases self-/polyreactivity, yielding increased tumor BCR reactivity for multiple CNS proteins, which likely contributes to CNS tropism of the lymphoma. The loss of MHC class I antigen expression supports tumor cell immune escape. Thus, specific and unique interactions of the tumor cells with resident CNS cells determine the hallmarks of PCNSL.
-
Altered DNA Methylation Profiles in SF3B1 Mutated CLL Patients
Mutations in splicing factor genes have a severe impact on the survival of cancer patients. Splicing factor 3b subunit 1 (SF3B1) is one of the most frequently mutated genes in chronic lymphocytic leukemia (CLL); patients carrying these mutations have a poor prognosis. Since the splicing machinery and the epigenome are closely interconnected, we investigated whether these alterations may affect the epigenomes of CLL patients. While an overall hypomethylation during CLL carcinogenesis has been observed, the interplay between the epigenetic stage of the originating B cells and SF3B1 mutations, and the subsequent effect of the mutations on methylation alterations in CLL, have not been investigated. We profiled the genome-wide DNA methylation patterns of 27 CLL patients with and without SF3B1 mutations and identified local decreases in methylation levels in SF3B1mut CLL patients at 67 genomic regions, mostly in proximity to telomeric regions. These differentially methylated regions (DMRs) were enriched in gene bodies of cancer-related signaling genes, e.g., NOTCH1, HTRA3, and BCL9L. In our study, SF3B1 mutations exclusively emerged in two out of three epigenetic stages of the originating B cells. However, not all the DMRs could be associated with the methylation programming of B cells during development, suggesting that mutations in SF3B1 cause additional epigenetic aberrations during carcinogenesis.
-
MARCKS affects cell motility and response to BTK inhibitors in CLL
Bruton tyrosine kinase (BTK) inhibitors are highly active drugs for the treatment of chronic lymphocytic leukemia (CLL). To understand the response to BTK inhibitors on a molecular level, we performed (phospho)proteomic analyses under ibrutinib treatment. We identified 3466 proteins and 9184 phosphopeptides (representing 2854 proteins) in CLL cells exhibiting a physiological ratio of phosphorylated serines (pS), threonines (pT), and tyrosines (pY) (pS:pT:pY). Expression of 83 proteins differed between unmutated immunoglobulin heavy-chain variable region (IGHV) CLL (UM-CLL) and mutated IGHV CLL (M-CLL). Strikingly, UM-CLL cells showed higher basal phosphorylation levels than M-CLL samples. Effects of ibrutinib on protein phosphorylation levels were stronger in UM-CLL, especially on phosphorylated tyrosines. The differentially regulated phosphopeptides and proteins clustered in pathways regulating cell migration, motility, cytoskeleton composition, and survival. One protein, myristoylated alanine-rich C-kinase substrate (MARCKS), showed striking differences in expression and phosphorylation level in UM-CLL vs M-CLL. MARCKS sequesters phosphatidylinositol-4,5-bisphosphate, thereby affecting central signaling pathways and clustering of the B-cell receptor (BCR). Genetically induced loss of MARCKS significantly increased AKT signaling and migratory capacity. CD40L stimulation increased expression of MARCKS. BCR stimulation induced phosphorylation of MARCKS, which was reduced by BTK inhibitors. In line with our in vitro findings, low MARCKS expression is associated with significantly higher treatment-induced leukocytosis and more pronounced decrease of nodal disease in patients with CLL treated with acalabrutinib.
-
Gene expression-based outcome prediction in advanced stage classical Hodgkin lymphoma treated with BEACOPP
-
Chemotherapy Induces Tumor-Associated Macrophages that Aid Adaptive Immune Responses in Ovarian Cancer
Neoadjuvant chemotherapy (NACT) may stimulate anticancer adaptive immune responses in high-grade serous ovarian cancer (HGSOC), but little is known about effects on innate immunity. Using omental biopsies from HGSOC, and omental tumors from orthotopic mouse HGSOC models that replicate the human tumor microenvironment, we studied the impact of platinum-based NACT on tumor-associated macrophages (TAM). We found that chemotherapy reduces markers associated with alternative macrophage activation while increasing expression of proinflammatory pathways, with evidence of inflammasome activation. Further evidence of a shift in TAM functions came from macrophage depletion via CSF1R inhibitors (CSF1Ri) in the mouse models. Although macrophage depletion in established disease had no impact on tumor weight or survival, CSF1Ri treatment after chemotherapy significantly decreased disease-free and overall survival. This decrease in survival was accompanied by significant inhibition of adaptive immune response pathways in the tumors. We conclude that chemotherapy skews the TAM population in HSGOC toward an antitumor phenotype that may aid adaptive immune responses, and therapies that enhance or sustain this during remission may delay relapse.
-
Whole-slide image analysis of the tumor microenvironment identifies low B-cell content as a predictor of adverse outcome in patients with advanced-stage classical Hodgkin lymphoma treated with BEACOPP
A subset of patients with advanced-stage classical Hodgkin Lymphoma (cHL) relapse or progress following standard treatment. Given their dismal prognosis, identifying this group of patients upfront represents an important medical need. While prior research has identified characteristics of the tumor microenvironment, which are associated with cHL outcomes, biomarkers that are developed and validated in this high-risk group are still missing. Here, we applied whole-slide image analysis (WSI), a quantitative, large-scale assessment of tumor composition that utilizes conventional histopathology slides. We conducted WSI on a study cohort with pre-treatment biopsies of 340 advanced-stage cHL patients enrolled in the HD12 and HD15 trials of the German Hodgkin Study Group (GHSG), and tested our results in in a validation cohort of 147 advanced-stage cHL patients within the GHSG HD18 trial. All patients were treated with BEACOPP-based regimens. By quantifying T cells, B cells, Hodgkin-Reed-Sternberg-cells and macrophages with WSI, 80% of all cells in the tumor tissue were identified. Crucially, low B cell count was associated with significantly reduced progression-free survival (PFS) and overall survival (OS), while T cell-, macrophage- and Hodgkin-Reed-Sternberg-cell content was not associated with the risk of progression or relapse in the study cohort. We further validated low B cell content as a prognostic factor of PFS and OS in the validation cohort and demonstrate good inter-observer agreement of WSI. WSI may represent a key tool for risk stratification of advanced-stage cHL that can easily be added to the standard diagnostic histopathology work-up.
-
Pericentromeric Satellite III transcripts induce etoposide resistance
Non-coding RNA from pericentromeric satellite repeats are involved in stress-dependent splicing processes, maintenance of heterochromatin, and are required to protect genome stability. Here we show that the long non-coding satellite III RNA (SatIII) generates resistance against the topoisomerase IIa (TOP2A) inhibitor etoposide in lung cancer. Because heat shock conditions (HS) protect cells against the toxicity of etoposide, and SatIII is significantly induced under HS, we hypothesized that the protective effect could be traced back to SatIII. Using genome methylation profiles of patient-derived xenograft mouse models we show that the epigenetic modification of the SatIII DNA locus and the resulting SatIII expression predict chemotherapy resistance. In response to stress, SatIII recruits TOP2A to nuclear stress bodies, which protects TOP2A from a complex formation with etoposide and results in decreased DNA damage after treatment. We show that BRD4 inhibitors reduce the expression of SatIII, restoring etoposide sensitivity.
-
Mutational mechanisms shaping the coding and noncoding genome of germinal center derived B-cell lymphomas
B cells have the unique property to somatically alter their immunoglobulin (IG) genes by V(D)J recombination, somatic hypermutation (SHM) and class-switch recombination (CSR). Aberrant targeting of these mechanisms is implicated in lymphomagenesis, but the mutational processes are poorly understood. By performing whole genome and transcriptome sequencing of 181 germinal center derived B-cell lymphomas (gcBCL) we identified distinct mutational signatures linked to SHM and CSR. We show that not only SHM, but presumably also CSR causes off-target mutations in non-IG genes. Kataegis clusters with high mutational density mainly affected early replicating regions and were enriched for SHM- and CSR-mediated off-target mutations. Moreover, they often co-occurred in loci physically interacting in the nucleus, suggesting that mutation hotspots promote increased mutation targeting of spatially co-localized loci (termed hypermutation by proxy). Only around 1% of somatic small variants were in protein coding sequences, but in about half of the driver genes, a contribution of B-cell specific mutational processes to their mutations was found. The B-cell-specific mutational processes contribute to both lymphoma initiation and intratumoral heterogeneity. Overall, we demonstrate that mutational processes involved in the development of gcBCL are more complex than previously appreciated, and that B cell-specific mutational processes contribute via diverse mechanisms to lymphomagenesis.
-
Regulation of MRE11A by UBQLN4 leads to cisplatin resistance in patients with esophageal squamous cell carcinoma
Resistance to standard cisplatin-based chemotherapies leads to worse survival outcomes for patients with esophageal squamous cell carcinoma (ESCC). Therefore, there is an urgent need to understand the aberrant mechanisms driving resistance in ESCC tumors. We hypothesized that ubiquilin-4 (UBQLN4), a protein that targets ubiquitinated proteins to the proteasome, regulates the expression of Meiotic Recombination 11 Homolog A (MRE11A), a critical component of the MRN complex and DNA damage repair pathways. Initially, immunohistochemistry analysis was conducted in specimens from patients with ESCC (n = 120). In endoscopic core ESCC biopsies taken from 61 patients who underwent neoadjuvant chemotherapy (NAC) (5-fluorouracil and cisplatin), low MRE11A and high UBQLN4 protein levels were associated with reduced pathological response to NAC (P < 0.001 and P < 0.001, respectively). Multivariable analysis of surgically resected ESCC tissues from 59 patients revealed low MRE11A and high UBLQN4 expression as independent factors that can predict shorter overall survival [P = 0.01, hazard ratio (HR) = 5.11, 95% confidence interval (CI), 1.45-18.03; P = 0.02, HR = 3.74, 95% CI, 1.19-11.76, respectively]. Suppression of MRE11A expression was associated with cisplatin resistance in ESCC cell lines. Additionally, MRE11A was found to be ubiquitinated after cisplatin treatment. We observed an amplification of UBQLN4 gene copy numbers and an increase in UBQLN4 protein levels in ESCC tissues. Binding of UBQLN4 to ubiquitinated-MRE11A increased MRE11A degradation, thereby regulating MRE11A protein levels following DNA damage and promoting cisplatin resistance. In summary, MRE11A and UBQLN4 protein levels can serve as predictors for NAC response and as prognostic markers in ESCC patients.
-
Ferroptosis response segregates small cell lung cancer (SCLC) neuroendocrine subtypes
Loss of TP53 and RB1 in treatment-naïve small cell lung cancer (SCLC) suggests selective pressure to inactivate cell death pathways prior to therapy. Yet, which of these pathways remain available in treatment-naïve SCLC is unknown. Here, through systemic analysis of cell death pathway availability in treatment-naïve SCLC, we identify non-neuroendocrine (NE) SCLC to be vulnerable to ferroptosis through subtype-specific lipidome remodeling. While NE SCLC is ferroptosis resistant, it acquires selective addiction to the TRX anti-oxidant pathway. In experimental settings of non-NE/NE intratumoral heterogeneity, non-NE or NE populations are selectively depleted by ferroptosis or TRX pathway inhibition, respectively. Preventing subtype plasticity observed under single pathway targeting, combined treatment kills established non-NE and NE tumors in xenografts, genetically engineered mouse models of SCLC and patient-derived cells, and identifies a patient subset with drastically improved overall survival. These findings reveal cell death pathway mining as a means to identify rational combination therapies for SCLC.
-
In-depth cell-free DNA sequencing reveals genomic landscape of Hodgkin’s lymphoma and facilitates ultrasensitive residual disease detection
Individualizing treatment is key to improve outcome and reduce long-term side-effects in any cancer. In Hodgkin lymphoma (HL), individualization of treatment is hindered by a lack of genomic characterization and technology for sensitive, molecular response assessment. Sequencing of cell-free (cf)DNA is a powerful strategy to understand an individual cancer genome and can be used to develop assays for extremely sensitive disease monitoring. In HL, a high proportion of cfDNA is tumor-derived making it a highly relevant disease model to study the role of cfDNA sequencing in cancer. Here, we introduce our targeted cfDNA sequencing platform and present the largest genomic landscape of HL to date, which was entirely derived by cfDNA sequencing. We comprehensively genotype and assess minimal residual disease in 324 samples from 121 patients, presenting an integrated landscape of mutations and copy number variations in HL. In addition, we perform a deep analysis of mutational processes driving HL, investigate the clonal structure of HL and link several genotypes to HL phenotypes and outcome. Finally, we show that minimal residual disease assessment by repeat cfDNA sequencing as early as a week after treatment initiation is feasible and predicts overall treatment response allowing highly improved treatment guidance and relapse prediction. Our study also serves as a blueprint showcasing the utility of our platform for other cancers with similar therapeutic challenges.
-
Active Akt signaling triggers CLL toward Richter transformation via overactivation of Notch1
Richter's transformation (RT) is an aggressive lymphoma that occurs upon progression from chronic lymphocytic leukemia (CLL). Transformation has been associated with genetic aberrations in the CLL phase involving TP53, CDKN2A, MYC, and NOTCH1; however, a significant proportion of RT cases lack CLL phase-associated events. Here, we report that high levels of AKT phosphorylation occur both in high-risk CLL patients harboring TP53 and NOTCH1 mutations as well as in patients with RT. Genetic overactivation of Akt in the murine Eµ-TCL1 CLL mouse model resulted in CLL transformation to RT with significantly reduced survival and an aggressive lymphoma phenotype. In the absence of recurrent mutations, we identified a profile of genomic aberrations intermediate between CLL and diffuse large B-cell lymphoma. Multiomics assessment by phosphoproteomic/proteomic and single-cell transcriptomic profiles of this Akt-induced murine RT revealed an S100 protein-defined subcluster of highly aggressive lymphoma cells that developed from CLL cells, through activation of Notch via Notch ligand expressed by T cells. Constitutively active Notch1 similarly induced RT of murine CLL. We identify Akt activation as an initiator of CLL transformation toward aggressive lymphoma by inducing Notch signaling between RT cells and microenvironmental T cells.
-
Constitutive activation of Lyn kinase enhances BCR responsiveness, but not the development of CLL in Eµ-TCL1 mice
The treatment of chronic lymphocytic leukemia (CLL) has been improved dramatically by inhibitors targeting B-cell receptor (BCR)-associated kinases. The tyrosine kinase Lyn is a key modulator of BCR signaling and shows increased expression and activity in CLL. To evaluate the functional relevance of Lyn for CLL, we generated a conditional knockin mouse model harboring a gain-of-function mutation of the Lyn gene (LynY508F), which was specifically expressed in the B-cell lineage (Lynup-B). Kinase activity profiling revealed an enhanced responsiveness to BCR stimulation in Lynup-B B cells. When crossing Lynup-B mice with Eµ-TCL1 mice (TCL1tg/wt), a transgenic mouse model for CLL, the resulting TCL1tg/wt Lynup-B mice showed no significant change of hepatomegaly, splenomegaly, bone marrow infiltration, or overall survival when compared with TCL1tg/wt mice. Our data also suggested that TCL1 expression has partially masked the effect of the Lynup-B mutation, because the BCR response was only slightly increased in TCL1tg/wt Lynup-B compared with TCL1tg/wt. In contrast, TCL1tg/wt Lynup-B were protected at various degrees against spontaneous apoptosis in vitro and upon treatment with kinase inhibitors targeting the BCR. Collectively, and consistent with our previous data in a Lyn-deficient CLL model, these data lend further suggest that an increased activation of Lyn kinase in B cells does not appear to be a major driver of leukemia progression and the level of increased BCR responsiveness induced by Lynup-B is insufficient to induce clear changes to CLL pathogenesis in vivo.
-
Ferroptotic pores induce Ca2+ fluxes and ESCRT-III activation to modulate cell death kinetics
Ferroptosis is an iron-dependent form of regulated necrosis associated with lipid peroxidation. Despite its key role in the inflammatory outcome of ferroptosis, little is known about the molecular events leading to the disruption of the plasma membrane during this type of cell death. Here we show that a sustained increase in cytosolic Ca2+ is a hallmark of ferroptosis that precedes complete bursting of the cell. We report that plasma membrane damage leading to ferroptosis is associated with membrane nanopores of a few nanometers in radius and that ferroptosis, but not lipid peroxidation, can be delayed by osmoprotectants. Importantly, Ca2+ fluxes during ferroptosis induce the activation of the ESCRT-III-dependent membrane repair machinery, which counterbalances the kinetics of cell death and modulates the immunological signature of ferroptosis. Our findings with ferroptosis provide a unifying concept that sustained increase of cytosolic Ca2+ prior to plasma membrane rupture is a common feature of regulated types of necrosis and position ESCRT-III activation as a general protective mechanism in these lytic cell death pathways.
-
Noncanonical effector functions of the T-memory-like T-PLL cell are shaped by cooperative TCL1A and TCR signaling
T-cell prolymphocytic leukemia (T-PLL) is a poor-prognostic neoplasm. Differentiation stage and immune-effector functions of the underlying tumor cell are insufficiently characterized. Constitutive activation of the T-cell leukemia 1A (TCL1A) oncogene distinguishes the (pre)leukemic cell from regular postthymic T cells. We assessed activation-response patterns of the T-PLL lymphocyte and interrogated the modulatory impact by TCL1A. Immunophenotypic and gene expression profiles revealed a unique spectrum of memory-type differentiation of T-PLL with predominant central-memory stages and frequent noncanonical patterns. Virtually all T-PLL expressed a T-cell receptor (TCR) and/or CD28-coreceptor without overrepresentation of specific TCR clonotypes. The highly activated leukemic cells also revealed losses of negative-regulatory TCR coreceptors (eg, CTLA4). TCR stimulation of T-PLL cells evoked higher-than-normal cell-cycle transition and profiles of cytokine release that resembled those of normal memory T cells. More activated phenotypes and higher TCL1A correlated with inferior clinical outcomes. TCL1A was linked to the marked resistance of T-PLL to activation- and FAS-induced cell death. Enforced TCL1A enhanced phospho-activation of TCR kinases, second-messenger generation, and JAK/STAT or NFAT transcriptional responses. This reduced the input thresholds for IL-2 secretion in a sensitizer-like fashion. Mice of TCL1A-initiated protracted T-PLL development resembled such features. When equipped with epitope-defined TCRs or chimeric antigen receptors, these Lckpr-hTCL1Atg T cells gained a leukemogenic growth advantage in scenarios of receptor stimulation. Overall, we propose a model of T-PLL pathogenesis in which TCL1A enhances TCR signals and drives the accumulation of death-resistant memory-type cells that use amplified low-level stimulatory input, and whose loss of negative coregulators additionally maintains their activated state. Treatment rationales are provided by combined interception in TCR and survival signaling.
-
CaMuS: simultaneous fitting and de novo imputation of cancer mutational signature
The identification of the mutational processes operating in tumour cells has implications for cancer diagnosis and therapy. These processes leave mutational patterns on the cancer genomes, which are referred to as mutational signatures. Recently, 81 mutational signatures have been inferred using computational algorithms on sequencing data of 23,879 samples. However, these published signatures may not always offer a comprehensive view on the biological processes underlying tumour types that are not included or underrepresented in the reference studies. To circumvent this problem, we designed CaMuS (Cancer Mutational Signatures) to construct de novo signatures while simultaneously fitting publicly available mutational signatures. Furthermore, we propose to estimate signature similarity by comparing probability distributions using the Hellinger distance. We applied CaMuS to infer signatures of mutational processes in poorly studied cancer types. We used whole genome sequencing data of 56 neuroblastoma, thus providing evidence for the versatility of CaMuS. Using simulated data, we compared the performance of CaMuS to sigfit, a recently developed algorithm with comparable inference functionalities. CaMuS and sigfit reconstructed the simulated datasets with similar accuracy; however two main features may argue for CaMuS over sigfit: (i) superior computational performance and (ii) a reliable parameter selection method to avoid spurious signatures.
-
Integrative Analysis of Pleomorphic Dermal Sarcomas Reveals Fibroblastic Differentiation and Susceptibility to Immunotherapy
Pleomorphic dermal sarcoma (PDS) is a rare malignant cutaneous tumor with an unknown cell of origin. Locally defined tumors can be treated by curative excisions, whereas advanced stages of the disease are difficult to treat, using standard regimens. We performed whole-exome sequencing on a cohort of 28 individuals and corresponding transcriptomic analysis on 21 patients, as well as quantitative IHC image analysis on 27 patients. PDS exhibits a universally high mutational load (42.7 mutations/mega base) with an inflamed, immunogenic tumor microenvironment. Three cases of PDS showed response to immune checkpoint blockade. Local mutation rate variation together with mRNA expression data demonstrate that PDS form a distinct entity, with PDGFRB as a lineage marker. In addition, we found that PDS is of mesenchymal, fibroblastic differentiation. PDS is of fibroblastic differentiation and exhibits a strong susceptibility to immunotherapy, including a high mutational burden and an inflamed tumor microenvironment.
-
Dynasore Blocks Ferroptosis through Combined Modulation of Iron Uptake and Inhibition of Mitochondrial Respiration
Ferroptosis is a form of regulated necrosis characterized by a chain-reaction of detrimental membrane lipid peroxidation following collapse of glutathione peroxidase 4 (Gpx4) activity. This lipid peroxidation is catalyzed by labile ferric iron. Therefore, iron import mediated via transferrin receptors and both, enzymatic and non-enzymatic iron-dependent radical formation are crucial prerequisites for the execution of ferroptosis. Intriguingly, the dynamin inhibitor dynasore, which has been shown to block transferrin receptor endocytosis, can protect from ischemia/reperfusion injury as well as neuronal cell death following spinal cord injury. Yet, it is unknown how dynasore exerts these cell death-protective effects. Using small interfering RNA suppression, lipid reactive oxygen species (ROS), iron tracers and bona fide inducers of ferroptosis, we find that dynasore treatment in lung adenocarcinoma and neuronal cell lines strongly protects these from ferroptosis. Surprisingly, while the dynasore targets dynamin 1 and 2 promote extracellular iron uptake, their silencing was not sufficient to block ferroptosis suggesting that this route of extracellular iron uptake is dispensable for acute induction of ferroptosis and dynasore must have an additional off-target activity mediating full ferroptosis protection. Instead, in intact cells, dynasore inhibited mitochondrial respiration and thereby mitochondrial ROS production which can feed into detrimental lipid peroxidation and ferroptotic cell death in the presence of labile iron. In addition, in cell free systems, dynasore showed radical scavenger properties and acted as a broadly active antioxidant which is superior to N-acetylcysteine (NAC) in blocking ferroptosis. Thus, dynasore can function as a highly active inhibitor of ROS-driven types of cell death via combined modulation of the iron pool and inhibition of general ROS by simultaneously blocking two routes required for ROS and lipid-ROS driven cell death, respectively. These data have important implications for the interpretation of studies observing tissue-protective effects of this dynamin inhibitor as well as raise awareness that off-target ROS scavenging activities of small molecules used to interrogate the ferroptosis pathway should be taken into consideration.
-
CHIP ubiquitylates NOXA and induces its lysosomal degradation in response to DNA damage
The BH3-only protein NOXA is a regulator of mitochondrial apoptosis by specifically antagonizing the anti-apoptotic protein MCL-1. Here we show that the E3 ubiquitin ligase CHIP controls NOXA stability after DNA damage. Our findings reveal that CHIP and MCL-1 are binding partners of NOXA and differentially define the fate of NOXA. Whereas NOXA is initially targeted to mitochondria upon MCL-1-binding, CHIP mediates ubiquitylation of cytosolic NOXA and promotes lysosomal degradation of NOXA, which is not bound by MCL-1. Our data indicate that MCL-1 defines NOXA abundance and its pro-apoptotic activity. Increased NOXA levels beyond this threshold are effectively removed by lysosomal protein degradation triggered via CHIP-mediated ubiquitylation. Together, these results shed new light on regulatory circuits controlling DNA damage response and identified the E3 ligase CHIP as a new molecular guardian, which restricts the cytosolic accumulation of NOXA upon genotoxic stress.
-
Death Receptors and Their Ligands in Inflammatory Disease and Cancer
On binding to their cognate ligands, death receptors can initiate a cascade of events that can result in two distinct outcomes: gene expression and cell death. The study of three different death receptor-ligand systems, the tumor necrosis factor (TNF)-TNF receptor 1 (TNFR1), the CD95L-CD95, and the TNF-related apoptosis-inducing ligand (TRAIL)-TRAIL-R1/2 system, has drawn the attention of generations of scientists over the past 50 years. This scientific journey, as often happens in science, has been anything but a straight line to success and discoveries in this field were often made by serendipity, catching the scientists by surprise. However, as Louis Pasteur pointed out, luck prefers the prepared mind. It is therefore not surprising that the most impactful discovery of the field to date, the fact that TNF inhibition serves as an effective treatment for several inflammatory and autoimmune diseases, has been like this. Luckily, the scientists who made this discovery were prepared and, most importantly, determined to harness their discovery for therapeutic benefit. Today's research on these death receptor-ligand systems has led to the discovery of a causal link between cell death induced by a variety of these systems and inflammation. In this review, we explain why we predict that therapeutic exploitation of this discovery may profoundly impact the future treatment of inflammatory disease and cancer.
-
Mitochondrial respiration controls neoangiogenesis during wound healing and tumour growth
The vasculature represents a highly plastic compartment, capable of switching from a quiescent to an active proliferative state during angiogenesis. Metabolic reprogramming in endothelial cells (ECs) thereby is crucial to cover the increasing cellular energy demand under growth conditions. Here we assess the impact of mitochondrial bioenergetics on neovascularisation, by deleting cox10 gene encoding an assembly factor of cytochrome c oxidase (COX) specifically in mouse ECs, providing a model for vasculature-restricted respiratory deficiency. We show that EC-specific cox10 ablation results in deficient vascular development causing embryonic lethality. In adult mice induction of EC-specific cox10 gene deletion produces no overt phenotype. However, the angiogenic capacity of COX-deficient ECs is severely compromised under energetically demanding conditions, as revealed by significantly delayed wound-healing and impaired tumour growth. We provide genetic evidence for a requirement of mitochondrial respiration in vascular endothelial cells for neoangiogenesis during development, tissue repair and cancer.
-
Dissecting intratumour heterogeneity of nodal B-cell lymphomas at the transcriptional, genetic and drug-response levels
Tumour heterogeneity encompasses both the malignant cells and their microenvironment. While heterogeneity between individual patients is known to affect the efficacy of cancer therapy, most personalized treatment approaches do not account for intratumour heterogeneity. We addressed this issue by studying the heterogeneity of nodal B-cell lymphomas by single-cell RNA-sequencing and transcriptome-informed flow cytometry. We identified transcriptionally distinct malignant subpopulations and compared their drug-response and genomic profiles. Malignant subpopulations from the same patient responded strikingly differently to anti-cancer drugs ex vivo, which recapitulated subpopulation-specific drug sensitivity during in vivo treatment. Infiltrating T cells represented the majority of non-malignant cells, whose gene-expression signatures were similar across all donors, whereas the frequencies of T-cell subsets varied significantly between the donors. Our data provide insights into the heterogeneity of nodal B-cell lymphomas and highlight the relevance of intratumour heterogeneity for personalized cancer therapy.
-
RIPK1‐mediated immunogenic cell death promotes anti‐tumour immunity against soft‐tissue sarcoma
Drugs that mobilise the immune system against cancer are dramatically improving care for many people. Dying cancer cells play an active role in inducing anti‐tumour immunity but not every form of death can elicit an immune response. Moreover, resistance to apoptosis is a major problem in cancer treatment and disease control. While the term “immunogenic cell death” is not fully defined, activation of receptor‐interacting serine/threonine‐protein kinase 1 (RIPK1) can induce a type of death that mobilises the immune system against cancer. However, no clinical treatment protocols have yet been established that would harness the immunogenic potential of RIPK1. Here, we report the first pre‐clinical application of an in vivo treatment protocol for soft‐tissue sarcoma that directly engages RIPK1‐mediated immunogenic cell death. We find that RIPK1‐mediated cell death significantly improves local disease control, increases activation of CD8+ T cells as well as NK cells, and enhances the survival benefit of immune checkpoint blockade. Our findings warrant a clinical trial to assess the survival benefit of RIPK1‐induced cell death in patients with advanced disease at limb extremities.
-
Meta-Analysis Reveals Significant Sex Differences in Chronic Lymphocytic Leukemia Progression in the Eµ-TCL1 Transgenic Mouse Model
The Eµ-TCL1 transgenic mouse model represents the most widely and extensively used animal model for chronic lymphocytic leukemia (CLL). In this report, we performed a meta-analysis of leukemia progression in over 300 individual Eµ-TCL1 transgenic mice and discovered a significantly accelerated disease progression in females compared to males. This difference is also reflected in an aggressive CLL mouse model with additional deletion of Tp53 besides the TCL1 transgene. Moreover, after serial adoptive transplantation of murine CLL cells, female recipients also succumbed to CLL earlier than male recipients. This sex-related disparity in the murine models is markedly contradictory to the human CLL condition. Thus, due to our observation we urge both careful consideration in the experimental design and accurate description of the Eµ-TCL1 transgenic cohorts in future studies.
-
Inhibition of Tumor VEGFR2 Induces Serine 897 EphA2-Dependent Tumor Cell Invasion and Metastasis in NSCLC
Anti-angiogenic treatment targeting vascular endothelial growth factor (VEGF)-VEGFR2 signaling has shown limited efficacy in lung cancer patients. Here, we demonstrate that inhibition of VEGFR2 in tumor cells, expressed in ∼20% of non-squamous non-small cell lung cancer (NSCLC) patients, leads to a pro-invasive phenotype. Drug-induced inhibition of tumor VEGFR2 interferes with the formation of the EphA2/VEGFR2 heterocomplex, thereby allowing RSK to interact with Serine 897 of EphA2. Inhibition of RSK decreases phosphorylation of Serine 897 EphA2. Selective genetic modeling of Serine 897 of EphA2 or inhibition of EphA2 abrogates the formation of metastases in vivo upon VEGFR2 inhibition. In summary, these findings demonstrate that VEGFR2-targeted therapy conditions VEGFR2-positive NSCLC to Serine 897 EphA2-dependent aggressive tumor growth and metastasis. These data shed light on the molecular mechanisms explaining the limited efficacy of VEGFR2-targeted anti-angiogenic treatment in lung cancer patients.
-
Richter transformation in chronic lymphocytic leukemia (CLL)-a pooled analysis of German CLL Study Group (GCLLSG) front line treatment trials
Richter transformation (RT) is defined as development of aggressive lymphoma in patients (pts) with CLL. The incidence rates of RT among pts with CLL range from 2 to 10%. The aim of this analysis is to report the frequency, characteristics and outcomes of pts with RT enrolled in trials of the GCLLSG. A total of 2975 pts with advanced CLL were reviewed for incidence of RT. Clinical, laboratory, and genetic data were pooled. Time-to-event data, starting from time of CLL diagnosis, of first-line therapy or of RT diagnosis, were analyzed by Kaplan-Meier methodology. One hundred and three pts developed RT (3%): 95 pts diffuse large B-cell lymphoma (92%) and eight pts Hodgkin lymphoma (8%). Median observation time was 53 months (interquartile range 38.1-69.5). Median OS from initial CLL diagnosis for pts without RT was 167 months vs 71 months for pts with RT (HR 2.64, CI 2.09-3.33). Median OS after diagnosis of RT was 9 months. Forty-seven pts (46%) received CHOP-like regimens for RT treatment. Three pts subsequently underwent allogeneic and two pts autologous stem cell transplantation. Our findings show that within a large cohort of GCLLSG trial participants, 3% of the pts developed RT after receiving first-line chemo- or chemoimmunotherapy. This dataset confirms the ongoing poor prognosis and high mortality associated with RT.
-
High efficacy of venetoclax plus obinutuzumab in patients with complex karyotype and chronic lymphocytic leukemia
-
The X-linked trichothiodystrophy-causing gene RNF113A links the spliceosome to cell survival upon DNA damage
Prolonged cell survival occurs through the expression of specific protein isoforms generated by alternate splicing of mRNA precursors in cancer cells. How alternate splicing regulates tumor development and resistance to targeted therapies in cancer remain poorly understood. Here we show that RNF113A, whose loss-of-function causes the X-linked trichothiodystrophy, is overexpressed in lung cancer and protects from Cisplatin-dependent cell death. RNF113A is a RNA-binding protein which regulates the splicing of multiple candidates involved in cell survival. RNF113A deficiency triggers cell death upon DNA damage through multiple mechanisms, including apoptosis via the destabilization of the prosurvival protein MCL-1, ferroptosis due to enhanced SAT1 expression, and increased production of ROS due to altered Noxa1 expression. RNF113A deficiency circumvents the resistance to Cisplatin and to BCL-2 inhibitors through the destabilization of MCL-1, which thus defines spliceosome inhibitors as a therapeutic approach to treat tumors showing acquired resistance to specific drugs due to MCL-1 stabilization.
-
Resistance Mechanisms to SYK Inhibition in Acute Myeloid Leukemia
Spleen tyrosine kinase (SYK) is a nonmutated therapeutic target in acute myeloid leukemia (AML). Attempts to exploit SYK therapeutically in AML have shown promising results in combination with chemotherapy, likely reflecting induced mechanisms of resistance to single-agent treatment in vivo. We conducted a genome-scale open reading frame (ORF) resistance screen and identified activation of the RAS-MAPK-ERK pathway as one major mechanism of resistance to SYK inhibitors. This finding was validated in AML cell lines with innate and acquired resistance to SYK inhibitors. Furthermore, patients with AML with select mutations activating these pathways displayed early resistance to SYK inhibition. To circumvent SYK inhibitor therapy resistance in AML, we demonstrate that a MEK and SYK inhibitor combination is synergistic in vitro and in vivo. Our data provide justification for use of ORF screening to identify resistance mechanisms to kinase inhibitor therapy in AML lacking distinct mutations and to direct novel combination-based strategies to abrogate these. SIGNIFICANCE: The integration of functional genomic screening with the study of mechanisms of intrinsic and acquired resistance in model systems and human patients identified resistance to SYK inhibitors through MAPK signaling in AML. The dual targeting of SYK and the MAPK pathway offers a combinatorial strategy to overcome this resistance.
-
The Evolutionary History of 2,658 Cancers
Cancer develops through a process of somatic evolution1,2. Sequencing data from a single biopsy represent a snapshot of this process that can reveal the timing of specific genomic aberrations and the changing influence of mutational processes3. Here, by whole-genome sequencing analysis of 2,658 cancers as part of the Pan-Cancer Analysis of Whole Genomes (PCAWG) Consortium of the International Cancer Genome Consortium (ICGC) and The Cancer Genome Atlas (TCGA)4, we reconstruct the life history and evolution of mutational processes and driver mutation sequences of 38 types of cancer. Early oncogenesis is characterized by mutations in a constrained set of driver genes, and specific copy number gains, such as trisomy 7 in glioblastoma and isochromosome 17q in medulloblastoma. The mutational spectrum changes significantly throughout tumour evolution in 40% of samples. A nearly fourfold diversification of driver genes and increased genomic instability are features of later stages. Copy number alterations often occur in mitotic crises, and lead to simultaneous gains of chromosomal segments. Timing analyses suggest that driver mutations often precede diagnosis by many years, if not decades. Together, these results determine the evolutionary trajectories of cancer, and highlight opportunities for early cancer detection.
-
Long Term Follow-up Data and Health-Related Quality of Life in Frontline Therapy of Fit Patients Treated With FCR Versus BR (CLL10 Trial of the GCLLSG)
Fludarabine, cyclophosphamide and rituximab (FCR) was compared to bendamustine and rituximab (BR) in an international, randomized, open label, phase 3 trial in 561 previously untreated, fit patients with chronic lymphocytic leukemia (CLL) without del (17p). Primary endpoint was progression free survival (PFS). The final primary endpoint analysis after 37.1 months median follow up failed to show the non-inferiority of BR as compared with FCR. With extended median follow up of 58.2 months, median PFS was 42.3 months in BR-treated patients versus 57.6 months for FCR-treated patients (Hazard Ratio [HR] 1.593; 95% CI 1.271–1.996; p < 0.0001). For patients > 65 years, median PFS was 48.5 months with BR versus 57.9 months with FCR without reaching statistical significance (HR 1.352; 95% CI 0.912–2.006; p = 0.134). Median OS was not reached for both arms with 5-year OS rates of 80.1% vs 80.9%, respectively (HR 1.108; 95% CI 0.755–1.627; p = 0.599). No statistically significant difference was found in the time to secondary malignancy between the 2 groups (at 5 years, 86.6% free from secondary malignancies in the BR group vs 83.8% in the FCR group [HR 0.801; 95% CI 0.507–1.267; p = 0.344]). In patients >65 years secondary neoplasia occurred more frequently after FCR treatment [28 of 86 (32.6%) patients] as compared with BR [18 of 107 (16.8%) patients; p = 0.011]. Health-related quality of life was similar in both treatments. Despite the improved PFS for FCR, OS did not differ. These results also suggest an increase in secondary neoplasia associated with FCR in elderly fit CLL patients.
-
Ferroptosis in Cancer Cell Biology
A major hallmark of cancer is successful evasion of regulated forms of cell death. Ferroptosis is a recently discovered type of regulated necrosis which, unlike apoptosis or necroptosis, is independent of caspase activity and receptor-interacting protein 1 (RIPK1) kinase activity. Instead, ferroptotic cells die following iron-dependent lipid peroxidation, a process which is antagonised by glutathione peroxidase 4 (GPX4) and ferroptosis suppressor protein 1 (FSP1). Importantly, tumour cells escaping other forms of cell death have been suggested to maintain or acquire sensitivity to ferroptosis. Therefore, therapeutic exploitation of ferroptosis in cancer has received increasing attention. Here, we systematically review current literature on ferroptosis signalling, cross-signalling to cellular metabolism in cancer and a potential role for ferroptosis in tumour suppression and tumour immunology. By summarising current findings on cell biology relevant to ferroptosis in cancer, we aim to point out new conceptual avenues for utilising ferroptosis in systemic treatment approaches for cancer.
-
An Autochthonous Mouse Model of Myd88- and BCL2-Driven Diffuse Large B-cell Lymphoma Reveals Actionable Molecular Vulnerabilities
Based on gene expression profiles, diffuse large B-cell lymphoma (DLBCL) is subdivided into germinal center B-cell–like (GCB) and activated B-cell–like (ABC) DLBCL. Two of the most common genomic aberrations in ABC-DLBCL are mutations in MYD88 as well as BCL2 copy-number gains. Here, we employ immune phenotyping, RNA sequencing, and whole-exome sequencing to characterize a Myd88- and BCL2-driven mouse model of ABC-DLBCL. We show that this model resembles features of human ABC-DLBCL. We further demonstrate an actionable dependence of our murine ABC-DLBCL model on BCL2. This BCL2 dependence was also detectable in human ABC-DLBCL cell lines. Moreover, human ABC-DLBCLs displayed increased PD-L1 expression compared with GCB-DLBCL. In vivo experiments in our ABC-DLBCL model showed that combined venetoclax and PD-1 blockade significantly increased the overall survival of lymphoma-bearing animals, indicating that this combination may be a viable option for selected human ABC-DLBCL cases harboring MYD88 and BCL2 aberrations.
-
Genomic analyses of PMBL reveal new drivers and mechanisms of sensitivity to PD-1 blockade
Primary mediastinal large B-cell lymphomas (PMBLs) are aggressive tumors that typically present as large mediastinal masses in young women. PMBLs share clinical, transcriptional, and molecular features with classical Hodgkin lymphoma (cHL), including constitutive activation of nuclear factor κB (NF-κB), JAK/STAT signaling, and programmed cell death protein 1 (PD-1)-mediated immune evasion. The demonstrated efficacy of PD-1 blockade in relapsed/refractory PMBLs led to recent approval by the US Food and Drug Administration and underscored the importance of characterizing targetable genetic vulnerabilities in this disease. Here, we report a comprehensive analysis of recurrent genetic alterations -somatic mutations, somatic copy number alterations, and structural variants-in a cohort of 37 newly diagnosed PMBLs. We identified a median of 9 genetic drivers per PMBL, including known and newly identified components of the JAK/STAT and NF-κB signaling pathways and frequent B2M alterations that limit major histocompatibility complex class I expression, as in cHL. PMBL also exhibited frequent, newly identified driver mutations in ZNF217 and an additional epigenetic modifier, EZH2. The majority of these alterations were clonal, which supports their role as early drivers. In PMBL, we identified several previously uncharacterized molecular features that may increase sensitivity to PD-1 blockade, including high tumor mutational burden, microsatellite instability, and an apolipoprotein B mRNA editing catalytic polypeptide-like (APOBEC) mutational signature. The shared genetic features between PMBL and cHL provide a framework for analyzing the mechanism of action of PD-1 blockade in these related lymphoid malignancies.
Link to the publication -
Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis
Caspase-8 is the initiator caspase of extrinsic apoptosis1,2 and inhibits necroptosis mediated by RIPK3 and MLKL. Accordingly, caspase-8 deficiency in mice causes embryonic lethality3, which can be rescued by deletion of either Ripk3 or Mlkl4-6. Here we show that the expression of enzymatically inactive CASP8(C362S) causes embryonic lethality in mice by inducing necroptosis and pyroptosis. Similar to Casp8-/- mice3,7, Casp8C362S/C362S mouse embryos died after endothelial cell necroptosis leading to cardiovascular defects. MLKL deficiency rescued the cardiovascular phenotype but unexpectedly caused perinatal lethality in Casp8C362S/C362S mice, indicating that CASP8(C362S) causes necroptosis-independent death at later stages of embryonic development. Specific loss of the catalytic activity of caspase-8 in intestinal epithelial cells induced intestinal inflammation similar to intestinal epithelial cell-specific Casp8 knockout mice8. Inhibition of necroptosis by additional deletion of Mlkl severely aggravated intestinal inflammation and caused premature lethality in Mlkl knockout mice with specific loss of caspase-8 catalytic activity in intestinal epithelial cells. Expression of CASP8(C362S) triggered the formation of ASC specks, activation of caspase-1 and secretion of IL-1β. Both embryonic lethality and premature death were completely rescued in Casp8C362S/C362SMlkl-/-Asc-/- or Casp8C362S/C362SMlkl-/-Casp1-/- mice, indicating that the activation of the inflammasome promotes CASP8(C362S)-mediated tissue pathology when necroptosis is blocked. Therefore, caspase-8 represents the molecular switch that controls apoptosis, necroptosis and pyroptosis, and prevents tissue damage during embryonic development and adulthood.
-
Residual abdominal lymphadenopathy after intensive frontline chemoimmunotherapy is associated with inferior outcome independently of minimal residual disease status in chronic lymphocytic leukemia
-
Quantitative analysis of super-resolved structures using ASAP
Super-resolution microscopy allows imaging of cellular structures with high throughput and detail. However, the efficient and quantitative analysis of images generated is challenging with existing tools. Here, we develop ASAP (automated structures analysis program) to enable rapid and automated detection, classification and quantification of super-resolved structures. We validate ASAP on ground truth data and demonstrate its broad applicability by analyzing images of nucleoporins, TORC1 complexes, endocytic vesicles and Bax pores.
-
Activated gp130 signaling selectively targets B cell differentiation to induce mature lymphoma and plasmacytoma
Aberrant activity of the glycoprotein 130 130/JAK/STAT3 (gp130/JAK/STAT3) signaling axis is a recurrent event in inflammation and cancer. In particular, it is associated with a wide range of hematological malignancies, including multiple myeloma and leukemia. Novel targeted therapies have only been successful for some subtypes of these malignancies, underlining the need for developing robust mouse models to better dissect the role of this pathway in specific tumorigenic processes. Here, we investigated the role of selective gp130/JAK/STAT3 activation by generating a conditional mouse model. This model targeted constitutively active, cell-autonomous gp130 activity to B cells, as well as to the entire hematopoietic system. We found that regardless of the timing of activation in B cells, constitutively active gp130 signaling resulted in the formation specifically of mature B cell lymphomas and plasma cell disorders with full penetrance, only with different latencies, where infiltrating CD138+ cells were a dominant feature in every tumor. Furthermore, constitutively active gp130 signaling in all adult hematopoietic cells also led to the development specifically of largely mature, aggressive B cell cancers, again with a high penetrance of CD138+ tumors. Importantly, gp130 activity abrogated the differentiation block induced by a B cell–targeted Myc transgene and resulted in a complete penetrance of the gp130-associated, CD138+, mature B cell lymphoma phenotype. Thus, gp130 signaling selectively provides a strong growth and differentiation advantage for mature B cells and directs lymphomagenesis specifically toward terminally differentiated B cell cancers.
-
Selective inactivation of hypomethylating agents by SAMHD1 provides a rationale for therapeutic stratification in AML
Hypomethylating agents decitabine and azacytidine are regarded as interchangeable in the treatment of acute myeloid leukemia (AML). However, their mechanisms of action remain incompletely understood, and predictive biomarkers for HMA efficacy are lacking. Here, we show that the bioactive metabolite decitabine triphosphate, but not azacytidine triphosphate, functions as activator and substrate of the triphosphohydrolase SAMHD1 and is subject to SAMHD1-mediated inactivation. Retrospective immunohistochemical analysis of bone marrow specimens from AML patients at diagnosis revealed that SAMHD1 expression in leukemic cells inversely correlates with clinical response to decitabine, but not to azacytidine. SAMHD1 ablation increases the antileukemic activity of decitabine in AML cell lines, primary leukemic blasts, and xenograft models. AML cells acquire resistance to decitabine partly by SAMHD1 up-regulation. Together, our data suggest that SAMHD1 is a biomarker for the stratified use of hypomethylating agents in AML patients and a potential target for the treatment of decitabine-resistant leukemia.
-
Venetoclax and Obinutuzumab in Patients with CLL and Coexisting Conditions
The BCL2 inhibitor venetoclax has shown activity in patients with chronic lymphocytic leukemia (CLL), but its efficacy in combination with other agents in patients with CLL and coexisting conditions is not known. In this open-label, phase 3 trial, we investigated fixed-duration treatment with venetoclax and obinutuzumab in patients with previously untreated CLL and coexisting conditions. Patients with a score of greater than 6 on the Cumulative Illness Rating Scale (scores range from 0 to 56, with higher scores indicating more impaired function of organ systems) or a calculated creatinine clearance of less than 70 ml per minute were randomly assigned to receive venetoclax–obinutuzumab or chlorambucil–obinutuzumab. The primary end point was investigator-assessed progression-free survival. The safety of each regimen was also evaluated. In total, 432 patients (median age, 72 years; median Cumulative Illness Rating Scale score, 8; median creatinine clearance, 66.4 ml per minute) underwent randomization, with 216 assigned to each group. After a median follow-up of 28.1 months, 30 primary end-point events (disease progression or death) had occurred in the venetoclax–obinutuzumab group and 77 had occurred in the chlorambucil–obinutuzumab group (hazard ratio, 0.35; 95% confidence interval [CI], 0.23 to 0.53; P<0.001). The Kaplan–Meier estimate of the percentage of patients with progression-free survival at 24 months was significantly higher in the venetoclax–obinutuzumab group than in the chlorambucil–obinutuzumab group: 88.2% (95% CI, 83.7 to 92.6) as compared with 64.1% (95% CI, 57.4 to 70.8). This benefit was also observed in patients with TP53 deletion, mutation, or both and in patients with unmutated immunoglobulin heavy-chain genes. Grade 3 or 4 neutropenia occurred in 52.8% of patients in the venetoclax–obinutuzumab group and in 48.1% of patients in the chlorambucil–obinutuzumab group, and grade 3 or 4 infections occurred in 17.5% and 15.0%, respectively. All-cause mortality was 9.3% in the venetoclax–obinutuzumab group and 7.9% in the chlorambucil–obinutuzumab group. These differences were not significant. Among patients with untreated CLL and coexisting conditions, venetoclax–obinutuzumab was associated with longer progression-free survival than chlorambucil–obinutuzumab. (Funded by F. Hoffmann–La Roche and AbbVie; ClinicalTrials.gov number, NCT02242942. opens in new tab.)
-
RIPK1 prevents TRADD-driven, but TNFR1 independent, apoptosis during development
RIPK1 is an essential downstream component of many pattern recognition and death receptors. RIPK1 can promote the activation of caspase-8 induced apoptosis and RIPK3-MLKL-mediated necroptosis, however, during development RIPK1 limits both forms of cell death. Accordingly, Ripk1-/- mice present with systemic cell death and consequent multi-organ inflammation, which is driven through the activation of both FADD-caspase-8 and RIPK3-MLKL signaling pathways causing perinatal lethality. TRADD is a death domain (DD) containing molecule that mediates signaling downstream of TNFR1 and the TLRs. Following the disassembly of the upstream receptor complexes either RIPK1 or TRADD can form a complex with FADD-caspase-8-cFLIP, via DD-DD interactions with FADD, facilitating the activation of caspase-8. We show that genetic deletion of Ripk1 licenses TRADD to complex with FADD-caspase-8 and activates caspase-8 during development. Deletion of Tradd provided no survival advantage to Ripk1-/- animals and yet was sufficient to reduce the systemic cell death and inflammation, rescue the intestinal and thymic histopathologies, reduce cleaved caspases in most tissues and rescue the anemia observed in Ripk1-/- neonates. Furthermore, deletion of Ripk3 is sufficient to rescue the neonatal lethality of Ripk1-/-Tradd-/- animals and delays but does not completely prevent early mortality. Although Ripk3 deletion provides a significant survival advantage, Ripk1-/-Tradd-/-Ripk3-/- animals die between 22 and 49 days, are runty compared to littermate controls and present with splenomegaly. These findings reveal a new mechanism by which RIPK1 limits apoptosis through blocking TRADD recruitment to FADD and preventing aberrant activation of caspase-8.
-
Cell Death and Inflammation - A Vital but Dangerous Liaison
The immune system has developed multiple ways to fight infection. Yet, it is constantly tasked with overcoming newly developing pathogenic mechanisms of resistance to host immunity. In most mammals, the stimulation of both innate and adaptive immune receptors can result in gene activation and cell death induction by apoptosis and necroptosis. RIPK1 and RIPK3 are key mediators of necroptosis; however, new findings support their role in the regulation of cell death-independent proinflammatory signaling. We discuss here the biological functions of RIPK1 and RIPK3, how they regulate cell death and inflammation, and the interplay between them. Finally, we discuss recent advances in our knowledge of linear ubiquitination which, alongside RIPK3 and caspase-8, exerts regulatory functions on RIPK1-mediated inflammation. Together, this review examines the complex interplay between RIPK1, RIPK3, and LUBAC that is important in regulating cell death and inflammatory signaling.
-
Extracellular vesicle measurements with nanoparticle tracking analysis - An accuracy and repeatability comparison between NanoSight NS300 and ZetaView
The expanding field of extracellular vesicle (EV) research needs reproducible and accurate methods to characterize single EVs. Nanoparticle Tracking Analysis (NTA) is commonly used to determine EV concentration and diameter. As the EV field is lacking methods to easily confirm and validate NTA data, questioning the reliability of measurements remains highly important. In this regard, a comparison addressing measurement quality between different NTA devices such as Malvern's NanoSight NS300 or Particle Metrix' ZetaView has not yet been conducted. To evaluate the accuracy and repeatability of size and concentration determinations of both devices, we employed comparative methods including transmission electron microscopy (TEM) and single particle interferometric reflectance imaging sensing (SP-IRIS) by ExoView. Multiple test measurements with nanospheres, liposomes and ultracentrifuged EVs from human serum and cell culture supernatant were performed. Additionally, serial dilutions and freeze-thaw cycle-dependent EV decrease were measured to determine the robustness of each system. Strikingly, NanoSight NS300 exhibited a 2.0-2.1-fold overestimation of polystyrene and silica nanosphere concentration. By measuring serial dilutions of EV samples, we demonstrated higher accuracy in concentration determination by ZetaView (% BIAS range: 2.7-8.5) in comparison with NanoSight NS300 (% BIAS range: 32.9-36.8). The concentration measurements by ZetaView were also more precise (% CV range: 0.0-4.7) than measurements by NanoSight NS300 (% CV range: 5.4-10.7). On the contrary, quantitative TEM imaging indicated more accurate EV sizing by NanoSight NS300 (% DTEM range: 79.5-134.3) compared to ZetaView (% DTEM range: 111.8-205.7), while being equally repeatable (NanoSight NS300% CV range: 0.8-6.7; ZetaView: 1.4-7.8). However, both devices failed to report a peak EV diameter below 60 nm compared to TEM and SP-IRIS. Taken together, NTA devices differ strongly in their hardware and software affecting measuring results. ZetaView provided a more accurate and repeatable depiction of EV concentration, whereas NanoSight NS300 supplied size measurements of higher resolution.
-
Venetoclax plus rituximab or obinutuzumab after allogeneic hematopoietic stem cell transplantation in chronic lymphocytic leukemia
For several decades, allogeneic hematopoietic stem cell transplantation (alloHCT) has been a therapeutic option in patients with chronic lymphocytic leukemia (CLL) with high-risk features such as del(17p) and/or TP53 mutations, complex aberrant karyotype, or chemotherapy-resistant disease. With the advent of new targeted agents, like ibrutinib, idelalisib, or venetoclax, an increasing number of patients with CLL is now managed without undergoing alloHCT.1,2 While alloHCT has curative potential, many patients with CLL are not eligible for transplant due to age and co-existing conditions, as the procedure can entail considerable treatment-related morbidity and mortality. Moreover, up to 50% of patients may experience relapsed disease after transplantation.
-
BCR-dependent lineage plasticity in mature B cells
B2 cells engage in classical antibody responses, whereas B1 cells are considered carriers of innate immunity, biased toward recognizing epitopes present on the surfaces of common pathogens and self antigens. To explore the role of B cell antigen receptor (BCR) specificity in driving B1 cell differentiation, we developed a transgenic system allowing us to change BCR specificity in B cells in an inducible and programmed manner. Mature B2 cells differentiated into bona fide B1 cells upon acquisition of a B1 cell-typical self-reactive BCR through a phase of proliferative expansion. Thus, B2 cells have B1 cell differentiation potential in addition to their classical capacity to differentiate into memory and plasma cells, and B1 differentiation can be instructed by BCR-mediated self-reactivity, in the absence of B1-lineage precommitment.
-
RIPK1 and Caspase-8 Ensure Chromosome Stability Independently of Their Role in Cell Death and Inflammation
Receptor-interacting protein kinase (RIPK) 1 functions as a key mediator of tissue homeostasis via formation of Caspase-8 activating ripoptosome complexes, positively and negatively regulating apoptosis, necroptosis, and inflammation. Here, we report an unanticipated cell-death- and inflammation-independent function of RIPK1 and Caspase-8, promoting faithful chromosome alignment in mitosis and thereby ensuring genome stability. We find that ripoptosome complexes progressively form as cells enter mitosis, peaking at metaphase and disassembling as cells exit mitosis. Genetic deletion and mitosis-specific inhibition of Ripk1 or Caspase-8 results in chromosome alignment defects independently of MLKL. We found that Polo-like kinase 1 (PLK1) is recruited into mitotic ripoptosomes, where PLK1's activity is controlled via RIPK1-dependent recruitment and Caspase-8-mediated cleavage. A fine balance of ripoptosome assembly is required as deregulated ripoptosome activity modulates PLK1-dependent phosphorylation of downstream effectors, such as BUBR1. Our data suggest that ripoptosome-mediated regulation of PLK1 contributes to faithful chromosome segregation during mitosis.
-
LUBAC determines chemotherapy resistance in squamous cell lung cancer
Lung squamous cell carcinoma (LSCC) and adenocarcinoma (LADC) are the most common lung cancer subtypes. Molecular targeted treatments have improved LADC patient survival but are largely ineffective in LSCC. The tumor suppressor FBW7 is commonly mutated or down-regulated in human LSCC, and oncogenic KRasG12D activation combined with Fbxw7 inactivation in mice (KF model) caused both LSCC and LADC. Lineage-tracing experiments showed that CC10+, but not basal, cells are the cells of origin of LSCC in KF mice. KF LSCC tumors recapitulated human LSCC resistance to cisplatin-based chemotherapy, and we identified LUBAC-mediated NF-κB signaling as a determinant of chemotherapy resistance in human and mouse. Inhibition of NF-κB activation using TAK1 or LUBAC inhibitors resensitized LSCC tumors to cisplatin, suggesting a future avenue for LSCC patient treatment.
-
New roles for B cell receptor associated kinases: when the B cell is not the target
Targeting of B cell receptor associated kinases (BAKs), such as Bruton’s tyrosine kinase (BTK) or phosphoinositol-3-kinase (PI3K) delta, by specific inhibitors has revolutionized the therapy of B lymphoid malignancies. BAKs are critical signaling transducers of BCR signaling and seem relevant in B cell lymphoma pathogenesis. The functional relevance of BTK for lymphoid malignancies is strongly supported by the observation that resistance to therapy in CLL patients treated with BTK inhibitors such as ibrutinib is often associated with mutations in genes coding for BTK or Phospholipase-C gamma (PLCɣ). In some contrast, next generation sequencing data show that BAKs are mutated at very low frequency in treatment-naïve B cell lymphomas. Therefore, it remains debatable whether BAKs are essential drivers for lymphoma development. In addition, results obtained by targeted deletion of BAKs such as Lyn and Btk in murine CLL models suggest that BAKs may be essential to shape the dialogue between malignant B cells and the tumor microenvironment (TME). Since BAKs are expressed in multiple cell types, BAK inhibitors may disrupt the lymphoma supportive microenvironment. This concept also explains the typical response to BAK inhibitor treatment, characterized by a long-lasting increase of peripheral blood lymphoid cells, due to a redistribution from the lymphoid homing compartments. In addition, BAK inhibitors have shown some efficacy in solid tumors, probably through mediator cells in the TME. This review summarizes and validates the evidence for BAK inhibitors being part of a class of agents that modulate the (hematopoietic) microenvironment of cancers.
-
UBQLN4 Represses Homologous Recombination and Is Overexpressed in Aggressive Tumors
Genomic instability can be a hallmark of both human genetic disease and cancer. We identify a deleterious UBQLN4 mutation in families with an autosomal recessive syndrome reminiscent of genome instability disorders. UBQLN4 deficiency leads to increased sensitivity to genotoxic stress and delayed DNA double-strand break (DSB) repair. The proteasomal shuttle factor UBQLN4 is phosphorylated by ATM and interacts with ubiquitylated MRE11 to mediate early steps of homologous recombination-mediated DSB repair (HRR). Loss of UBQLN4 leads to chromatin retention of MRE11, promoting non-physiological HRR activity in vitro and in vivo. Conversely, UBQLN4 overexpression represses HRR and favors non-homologous end joining. Moreover, we find UBQLN4 overexpressed in aggressive tumors. In line with an HRR defect in these tumors, UBQLN4 overexpression is associated with PARP1 inhibitor sensitivity. UBQLN4 therefore curtails HRR activity through removal of MRE11 from damaged chromatin and thus offers a therapeutic window for PARP1 inhibitor treatment in UBQLN4-overexpressing tumors.
Upcoming Events
-
10:00 :
Early Career Researchers in Oncology: funding opportunities
-
08:00 :
West German Lymphoma Symposium
-
13:00 :
SFB 1530 Seminar Series: Florian Bassermann
-
13:00 :
SFB 1530 Seminar Series: Sebastian Kobold
-
13:00 :
SFB 1530 Seminar Series: Ulrike Köhl
-
14:30 :
Annual Retreat
-
09:00 :
Annual Retreat
-
13:00 :
SFB 1530 Seminar Series: Luca Scorrano
-
09:00 :
SFB 1530 Symposium
Principal Investigators

























